杰出的分子设计软件
创新科技,直观软件
概述
Cresset应用程序基于坚实的科学,提供您可信赖的结果。我们的目标是帮助您发现、设计和优化最好的分子。我们相信计算方法能帮助我们更好地理解化学结构和蛋白质的性质和行为。优秀的科学是我们所做的一切的核心,我们总是优先考虑科学的严谨性而不是易用性和更快的运行时间。
Cresset技术简介
Cresset技术集中于XED力场在设计新的小分子生物活性化合物中的应用。XED方法通过复杂的原子描述来模拟远离原子中心的电荷,从而改进了传统的分子力学。这使得能够更详细地描述静电并优异地重现分子间相互作用。XED力场由Andy Vinter博士开发,在Cresset改进,它正确地模拟了取代基对芳香族化合物的效应、复杂芳香族化合物中电荷密度的变化以及小分子、水和蛋白质之间的分子间相互作用。
用XED力场计算静电
分子识别的最重要影响因素是静电,但它也受形状和疏水性的影响。Cresset方法将配体或蛋白质周围的静电环境描述为分子相互作用电势(MIP)或场。MIP可以描述配体与蛋白间的所有能量上重要的相互作用、用于观察MIP或蛋白质,提供了为什么一些配体比其它配体结合更强的清楚见解。用静电而不是结构来描述分子使我们能够灵活地比较不同系列的分子。
静电相似性的计算用于小分子的发现
Cresset拥有比较两个分子间相互作用势并计算相似性的专利方法。场相似性可用于:
- 设计新的分子
- 骨架跃迁在化学空间中发现新的区域
- 理解并破译SAR
- 找到新的先导化合物结构
XED力场
Cresset聚焦于用静电来描述分子。为了使其有效,静电模型必须是精确的。量子力学计算可以给出非常精确的静电势,但是在大多数情况下仍然太慢。因此,需要一种在分子力学环境中计算静电势的精确方法。
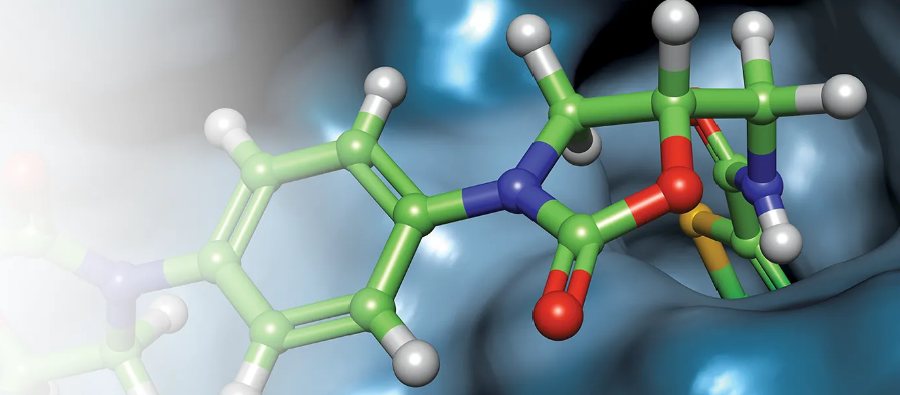
大多数标准力场使用原子中心电荷(ACC)近似来处理:分子的静电由放置在原子核上的一组点偏电荷(point partial charge)来近似处理。计算这些偏电荷的方法很多(Gasteiger-Hückel,AM1-BCC等), 但基本模型是每个原子一个点电荷。该方法可以很好地处理总的长程静电势(例如偶极矩),但是在描述分子表面附近的静电势时表现较差。这是因为原子不是带电的球体:它们有孤对电子,π 轨道,Sigma洞(Sigma hole)等等。此外,原子和分子是可极化的,并且对外部电场产生响应并改变静电行为。ACC模型不包括这些影响中的任何一个。诸如AMOEBA之类的较新的力场通过在原子上放置显式多极和极化函数来解决这个问题,这确实给出了更真实的静电势。虽然这些力场在蛋白质上表现良好,但是存在参数可迁移的问题,这使得它们在很大程度上不适合于配体的模拟。
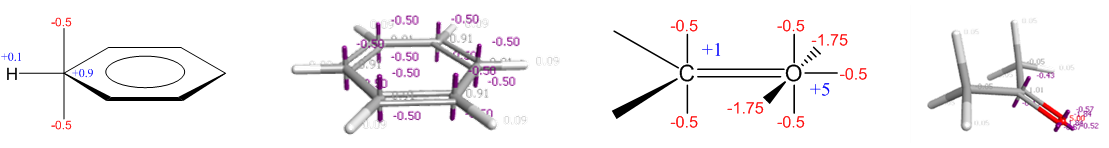
苯与丙酮的XED模型及其外加的点电荷
XED力场是解决这些静电问题的第一个主要尝试者,它并不是通过在原子上放置显式多极(explicit multipoles),而是通过在原子周围放置额外的单极(monopoles)。该技术最初由Hunter和Sanders[1]引入以模拟芳香-芳香相互作用,并由Cresset的创始人Andy Vinter(JCAMD 1994)[2]扩展到完全通用的力场中。
附加单极点或XEDS(扩展电子分布),在力场内被处理为具有零范德华半径的原子。它们没有被放置在相对于它们的母原子的刚性几何形状中。相反,它们具有键拉伸和角弯曲电势,并且可以在外部(和分子内)静电电势的影响下移动,从而允许对极化率进行直接建模。更复杂的内部静电模型允许在不引入特定扭转参数的情况下对分子内静电/轨道相互作用(例如,同系物效应)进行建模:同系物效应自然地落在静电模型之外,并且不需要在之后添加。
已经证明XED力场为芳香-芳香相互作用的能量提供了优异的定量结果。Cresset的学术合作者使用XED模型来研究和预测特定的分子间相互作用。例如,Chris Hunter教授使用NMR和计算化学(取代基对芳族堆叠相互作用的影响)研究了一系列二芳基酰胺的二聚体。他发现XED模型精确地预测了实验观察到的结合常数。

二芳基酰胺二聚体(左)与实验观测到的结合常数
芳香族相互作用的优良模型用于设计连接胶原模拟纤维的芳族”拉链”(血栓形成的胶原模拟肽:由疏水性相互作用驱动的三重螺旋基纤维的自组装)。
XED力场提供了苯基和五氟苯基相互作用定性和定量的正确结果
在过去20年中XED力场经历了多次改进。与大多数其它力场不同,XED力场尽可能地用实验数据(微波构象能量、小分子晶体结构等)进行参数化,而不是完全依赖于从头计算。2012年发布的XED3提供了对氮的改进处理,以及许多其他方面的增强。XED 3力场不再需要为三角形或四面体氮分配单独的原子类型,而是在计算中确定适合于任何给定分子环境的金字塔化程度,从而允许从完全平坦的N到完全金字塔的N的连续体。此外,XED 3改进了对卤素的描述,正确地描述了较重卤素中的”σ 孔”(Sigma hole)并给出了良好的卤素键合结果。Cresset将持续在此基础上进行开发和改进力场。
计算场并用于评估分子间相互作用
Cresset的3D配体相似性技术基础是:用分子静电相互作用电势(molecular electrostatic interaction potentials,MIPs)或场(Field)来比较分子。分子的MIP是标量场,空间中每个点的值是带电探针原子(具有范德华参数的氧)与分子的相互作用能,是使用XED力场计算的。由于分子内部的相互作用能定义得很差,所以在范德华相互作用能为正且大于静电能绝对值的任何地方将场值设置为零。
处理全部的3D标量势在计算上是很困难的。您可以采集网格上的值,但是会遇到测量尺度差异(Gauge variance)以网格间距等问题从而导致不可再现性。相反,Cresset所做的是询问”最大/最小场在哪里?”,每个这样的极值被称为“场点”(Field point),并且对于任何给定的分子构象,场点的集合是唯一的。场点通常被显示为彩色球体,其中每个场点的可视范围由场的大小确定,场越强则球越大。这让你一眼就能看到分子在哪里可以与另一个分子产生局部最大的静电作用。论文《Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation》[3]详细介绍了场的定义和计算场点位置的算法。
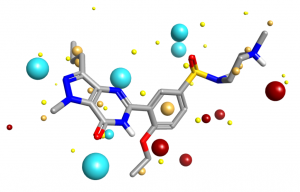
从PDB 1UDT提取出的西地那非的分子场点
作为XED力场开发的一部分Cresset的场已被验证过,并且一直与小分子晶体结构的实验数据进行了广泛地比较。氢键供体和受体在官能团周围的分布是相互作用势的良好代表,并且计算获得的场点模式与该信息一致。场对于描述芳香体系的性质也是特别有用的(建立在XED力场对这些性质极好描述的基础上):芳香环周围的场表面含有大量的信息,包括电子的富/缺信息、电荷密度的排列方式,π-堆积相互作用的强度等等。下图展示了几个环的实验数据。

Isostar plot of oxazole, pyridine, fluorobenzene
无论何时,正确地处理形式电荷状态对分子的静电描述是至关重要的。 Cresset有一套复杂的基于规则的系统用于分配形式电荷,虽然不是完整的pKa预测器,但可以正确地给绝大多数类药分子分配在pH 7时的质子化状态。然而,仅仅只是分配正确的形式电荷状态是不够的。比起中性分子,对离子来说溶剂化要重要得多,因此需要做更多的努力来解决这个问题。我们处理小分子形式电荷的完整算法在博客文章《如何计算带电分子周围的静电环境》中有详细介绍。
蛋白的场
Cresset的场和场点在小分子结构上得到了广泛的验证。将该方法扩展到蛋白则更加困难,因为需要更多地关注体系准备、电荷状态和溶剂化效应。 Flare,我们基于结构的药物设计平台,解决了这些问题,特别是通过仔细的蛋白结构准备和基于Mehler改进介电函数[4]的使用。
所得的相互作用势(PIP,protein interaction potential,以便将它们与配体MIP区分开)对于分析蛋白质活性位点和确定配体上何处需要改变性质以实现最佳结合是极其有用的-参见算例《比较Btk抑制剂的配体和蛋白静电势》。

配体4L6与4Z3V蛋白相互作用势的叠加。左上:”干”的活性位点,不包括结晶水分子。右上方:”湿”的活性位点,包括稳定的水分子。底部:配体4L6的场。绘图方法:蛋白质相互作用势等值图isolevel=3; 配体场的等值图isolevel =2。
场相似性用于基于配体的设计
高质量的静电势可用这种电荷结构的高级表示来计算,接着就可以用来比较两个构象间的静电相似性。你可以通过比较两个分子的场点来做到这一点,这就产生了一种类似药效团的技术。然而,更好的解决方案是考虑全部的静电势。

完整的静电势包含的信息不仅只是氮原子是个受体这个事实
相似性需要根据潜在的静电势来计算,而不仅仅是根据场点来计算。虽然计算困难,但Cresset优雅、高效地解决了这个问题。仅在其中一个构象具有场点的地方比较两个个分子的场; 让场计算的数量有限,但是必须确保至少一个构象表明场该是重要的地方(即,在场点处)被计算。完整的算法见Cheeseright等人的文献(JCIM 2006)[3]。

通过在A的场点所在的位置确定B的场势来计算构象A到构象B的分数。还可以通过计算相反的B到A分数并对两者取平均值的进行对称操作获得综合打分。然而,仅仅对特定的叠合方式进行评分是不够的:您需要能够找到最佳的叠合方式。这是一个困难的全局优化问题。Cresset的解决方案是通过计算两个构象的场点集合之间的匹配来生成一套初始的叠合:匹配考虑场点类型和所有的场点间距离匹配(在距离容限内)。每个匹配生成一个叠合 (通过3D场点匹配的最小二乘拟合),再根据上述场相似性算法对叠合进行打分。然后,将打分最高的叠合视为这两个构象的”正确”叠合。
当然,在许多情况下,不知道分子应该处于哪种构象。如果比较两种已知的生物活性构象,例如来自蛋白晶体结构,则可以直接用场相似性算法。更常见的情况是其中一个分子具有未知的构象:最好的例子是虚拟筛选-利用查询分子的3D构象进行搜索,但是不知道被搜索分子的先验构象是否将是恰当。在这种情况下,进行构象搜索,产生一套表示分子的可及构象空间的构象集。它们中的每一个都与查询分子进行叠合,并将得分最好的叠合作为分子的整体得分。
在某些情况下,有必要在不知道任何分子的生物活性构象的情况下比较它们。在这种情况下,计算两个分子的构象分布,并将第一个分子的每一个构象与第二个分子的每一个构象进行比较。这也是我们FieldTemplatter技术进行药效团识别的流程。
场相似性算法是一种快速算法。将一个分子的查询构象与另一个分子的100个构象比较在单个CPU(1核心)上花费大约1-2秒。这已被证明是非常有效的虚拟筛选。对Cresset相似性算法的性能评估表明,我们的Blaze虚拟筛选软件的性能显著优于分子对接技术(Cheeseright等人的JCIM 2008)[5]。该算法可以通过将其与形状相似性计算(Grant等人)组合来增强:总体相似性是场相似性和形状相似度的加权组合。在大多数情况下,使用相等的权重(50%形状,50%场),但这可由用户针对特定情况自行设定。
场相似性计算的增强包括添加场约束、药效团约束和排除体积。场约束用于将场的特定区域标记为比其它区域更重要,药效团约束要求特定类型的原子彼此接近,而排除体积能够利用蛋白结构信息将叠合限制于可及的空间内。所有的这些都显著地提高叠合和打分的精度。
静电互补(Electrostatic Complementarity,EC)
Cresset将配体静电的分析与蛋白静电的分析结合起来,以产生配体-蛋白质复合物的静电互补性(EC)的视觉评估和数值评估。 Cresset EC方法在计算上是廉价的,并且可以应用于大的数据集。通过将该方法应用于一系列文献数据集,我们分析了视觉和数值分量,表明它与报道的生物活性差异相关,并能够预测报道的生物活性差异–参见我们在J.Med Chem 2019[6]中的论文。
EC分析也已应用于激酶选择性预测、链霉亲和素突变分析和蛋白质环境中配体质子化状态的选择。本工作的结果表明,评估EC可以是用于分析和优化蛋白-配体静电相互作用的有力工具,使得量化静电驱动SAR和预测静电靶标选择性成为可能。
下图展示了生物素-链霉抗生物素蛋白复合物在配体和蛋白SAS上的ESP和EC,显示了蛋白和配体的正和负静电势区域的密切匹配如何导致良好的EC。

下面,我们计算EC分值,它对去溶剂化效应做了近似校正,并且允许EC在蛋白质或配体溶剂可及表面上的局部可视化,其中积分在配体SAS、ESPL和ESPP、配体和蛋白质ESP值以及MAX(ESPL, ESPP, k)上,蛋白质或配体ESP值与零或常数k的偏差较大,则ESP越大。配体和蛋白质ESP值被封端到观察到的水分子的最大ESP值,以近似校正去溶剂化。 EC分数范围从1到-1,分别对应于完美的EC或完美的静电冲突。由于配体的溶剂暴露部分贡献较少的蛋白-配体复合物的静电互补性的信息,距离任何白质原子大于3Å的配体SAS区域通过距离依赖性因子按比例缩小。
Cresset蛋白-配体EC是通过比较生成的配体或蛋白质溶剂可及表面(SAS)的所有顶点处的蛋白和配体的静电势(ESP)值来计算的 [7,8]。完美的静电互补性意味着在每个顶点处配体ESP值与具有相反符号的相同量值的蛋白质ESP值配对。
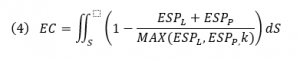
Cresset EC方法已应用于已报道的XIAP-BIR3数据集,结果表明该方法可检测并对导致生物活性改变的XIAP配体中的静电差异进行定量。参见《Investigating the SAR of XIAP ligands with Electrostatic Complementarity maps and scores》。
炼金术FEP计算结合自由能(FEP)
自由能方法作为一种计算配体与蛋白结合的绝对或相对自由能的方法,在药物发现中的应用越来越广泛。通过模拟水盒中的蛋白和配体,可以捕获相互作用的动力学,从而提供比更便宜的方法(如分子对接打分函数或MM/GBSA)更加准确的结合能估计。得益于误差消除,一般来说相对自由能估计明显比绝对自由能估计更容易计算且更可靠。
炼金术法自由能计算化合物A相对于参比化合物B的结合能涉及在溶剂中蛋白环境下将化合物A转化为B的热力学循环(图1)。
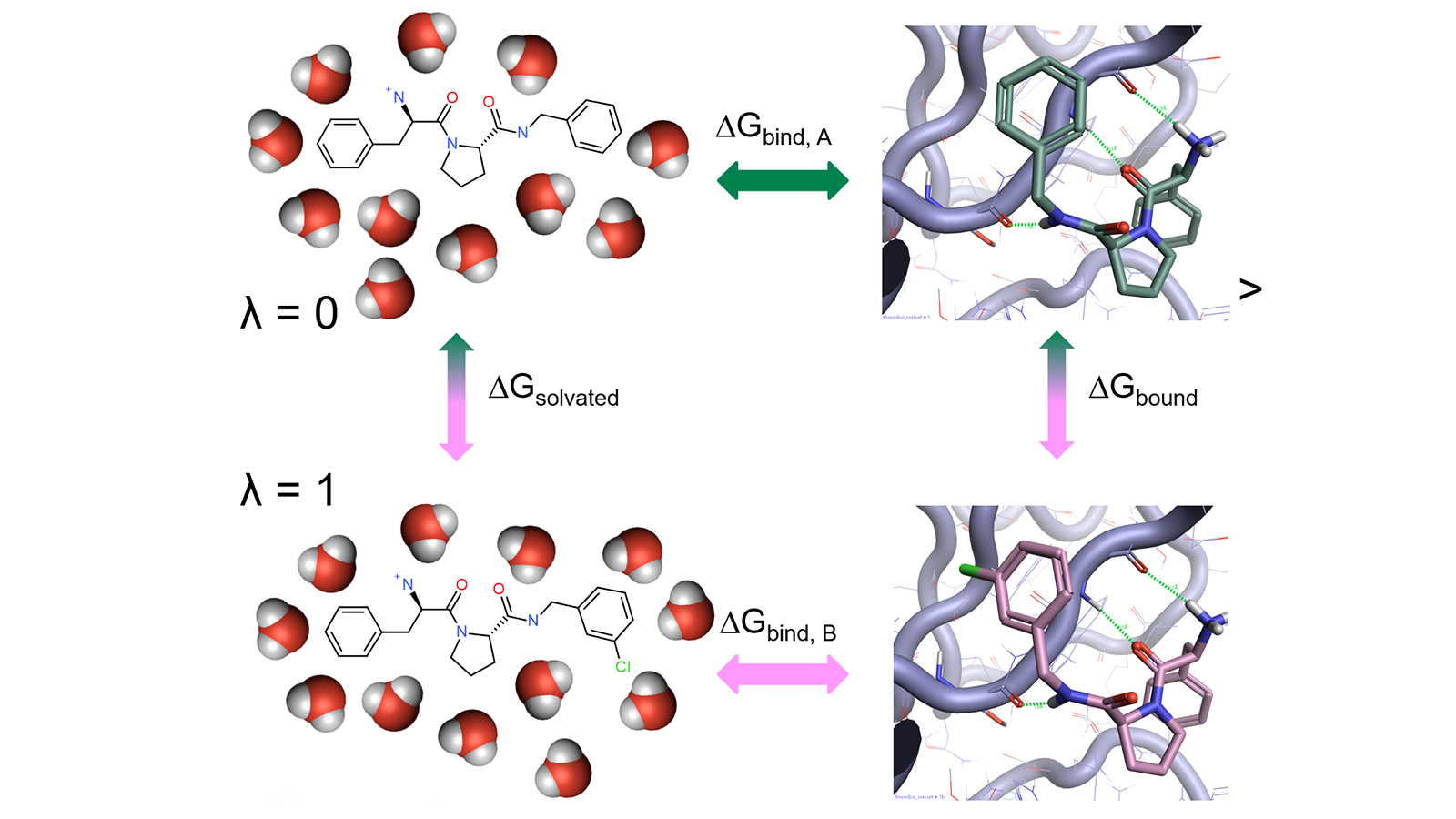
图1. 相对自由能计算中的热力学循环
A 和 B 的对应状态需要在它们的势能分布上有显着的重叠,否则为 A 采样的构型对于 B 的概率可能很低,这导致自由能变化的收敛缓慢,甚至不能获得计算结果。通常通过引入耦合参数λ来克服此限制,该参数取0到1之间的值,以插入到终态的势能函数中。然后通过不同数量的人工(“炼金术”)状态实现从A到B的转换,这些状态以中间λ值(所谓的λ窗口)为特征,改善了与相邻状态之间势能分布的重叠(图2)。
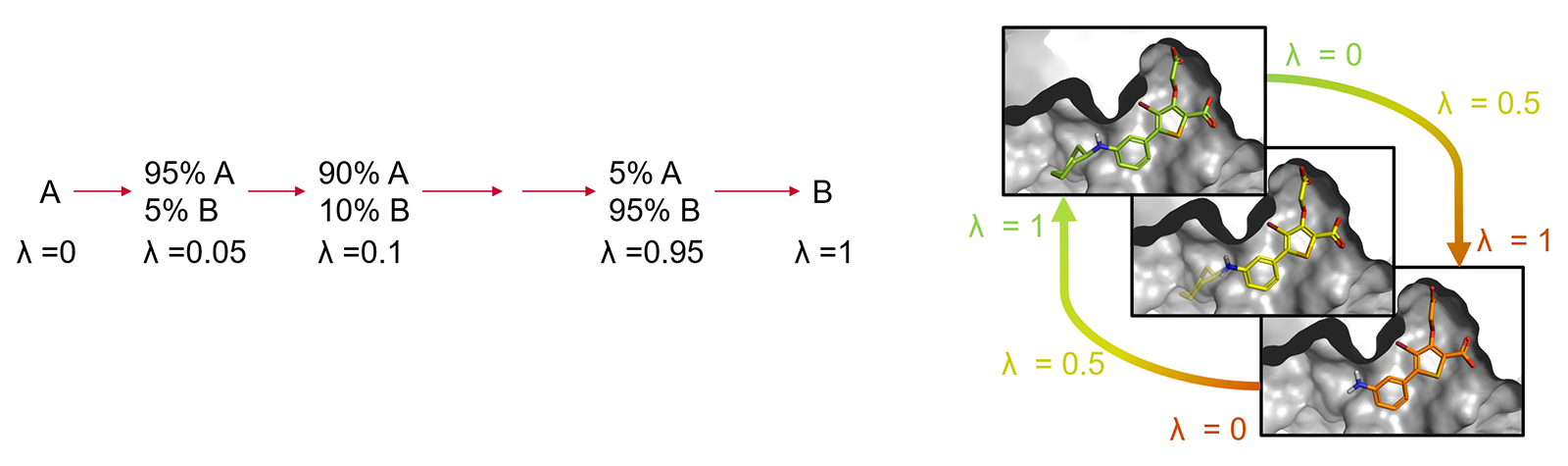
图2. 配体A到B的炼金术转化经由一系列非物理中间体结构
Cresset 相对自由能计算的实现基于一套开源工具(图 3),特别是 SIRE 工具包的 SOMD 模块。SOMD 包已成功应用于一系列片段、类药物小分子、碳水化合物和主-客体系统的相对和绝对结合自由能研究。

图3. Flare FEP用到的开源工具
Flare FEP用Wang 等人9的基准数据集进行测试,结果如图4、5所示。关于Flare FEP方法以及如何执行此基准测试的更多详细信息,请参阅我们的论文10。
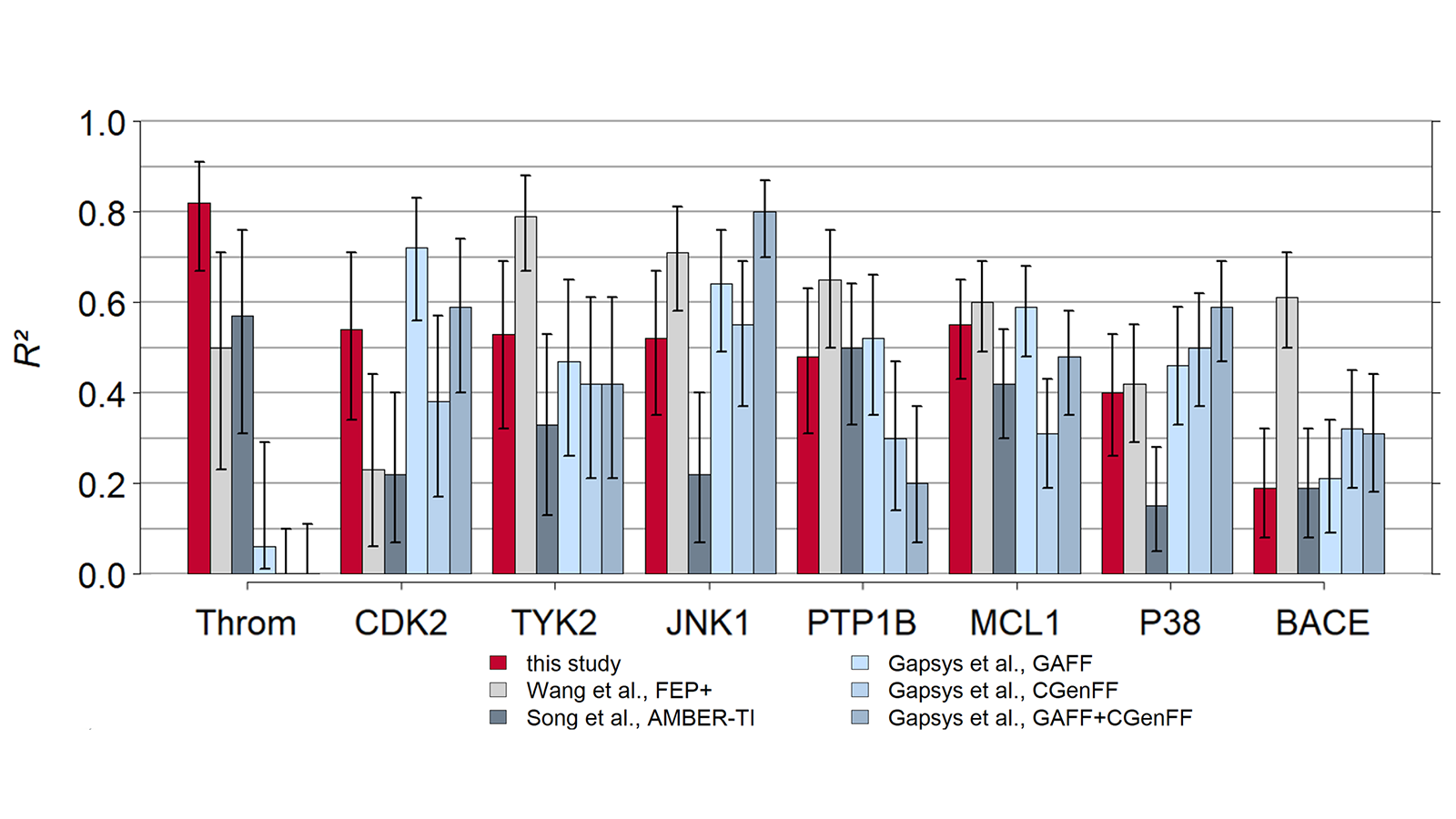
图4. 预测值-实验值的相关性
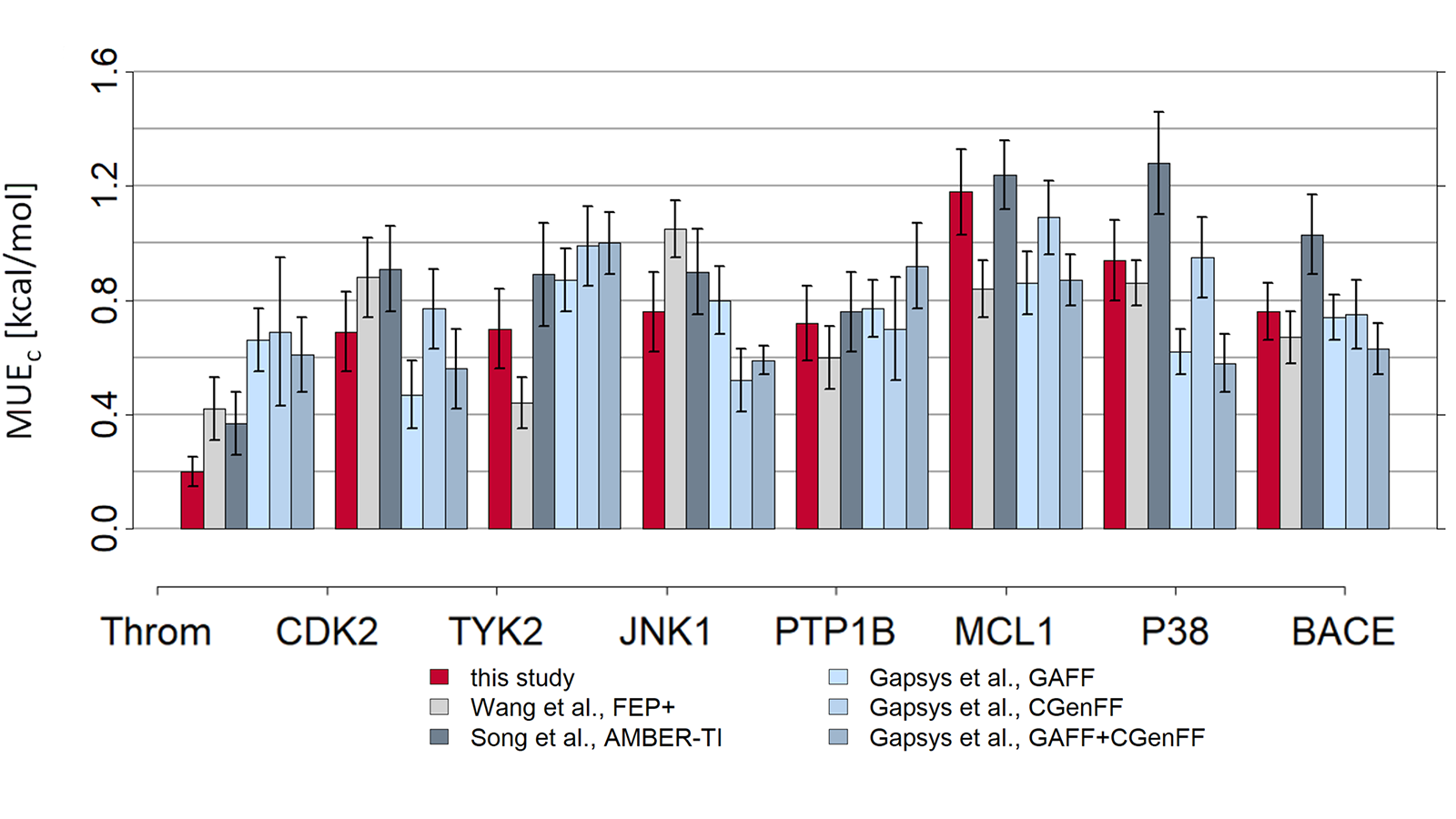
图5. 预测值-实验值的MUE
设置FEP计算最复杂的部分之一是创建网络(拓扑构造)。由于FEP方法在计算密切相关分子的相对活性时最可靠和准确,因此谨慎设置网络非常重要。理想情况下,每个分子应该通过至少两个连接(以提供冗余)连接到网络的其余部分,这些连接应该是密切相关的分子,所有分子都应该在一个小循环中,以便可以进行循环闭合分析获得误差,并且图的整体直径不能太大,以避免从图的一侧到另一侧的系统误差。Flare使用 LOMAP工具 (https://github.com/MobleyLab/Lomap) 的修改版本来执行此分析,从而生成如图6所示的图。LOMAP程序已经过优化,使其打分与我们FEP方法的已知行为保持一致,允许3D坐标用于原子映射,并正确处理分子之间的手性反转。
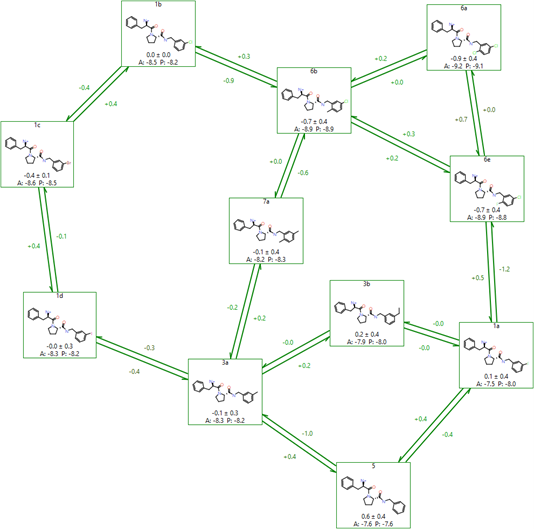
图6. 在Flare中LOMAP创建FEP网络的例子
Flare V5是Flare的最新版本,已经对FEP 方法进行了一些重大改进。首先,系统设置和积分器的改进使我们能够将计算速度比Flare V4(图 7)中的等效计算速度提高 2.5-3 倍。
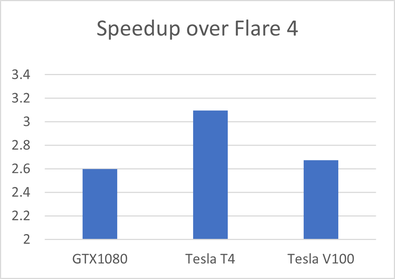
图7. Flare V5比Flare V4在计算速度上的提升
此外,还开发了一种算法,可以使用一个很短的预计算自动确定每个链接 λ 值的最佳规划(图 8)。此过程允许获得一组 λ 值,在最小化计算次数的同时,保持模拟之间良好的相位空间重叠。 在测试中,这通常会导致需要执行的 λ 窗口模拟数量减少30%。
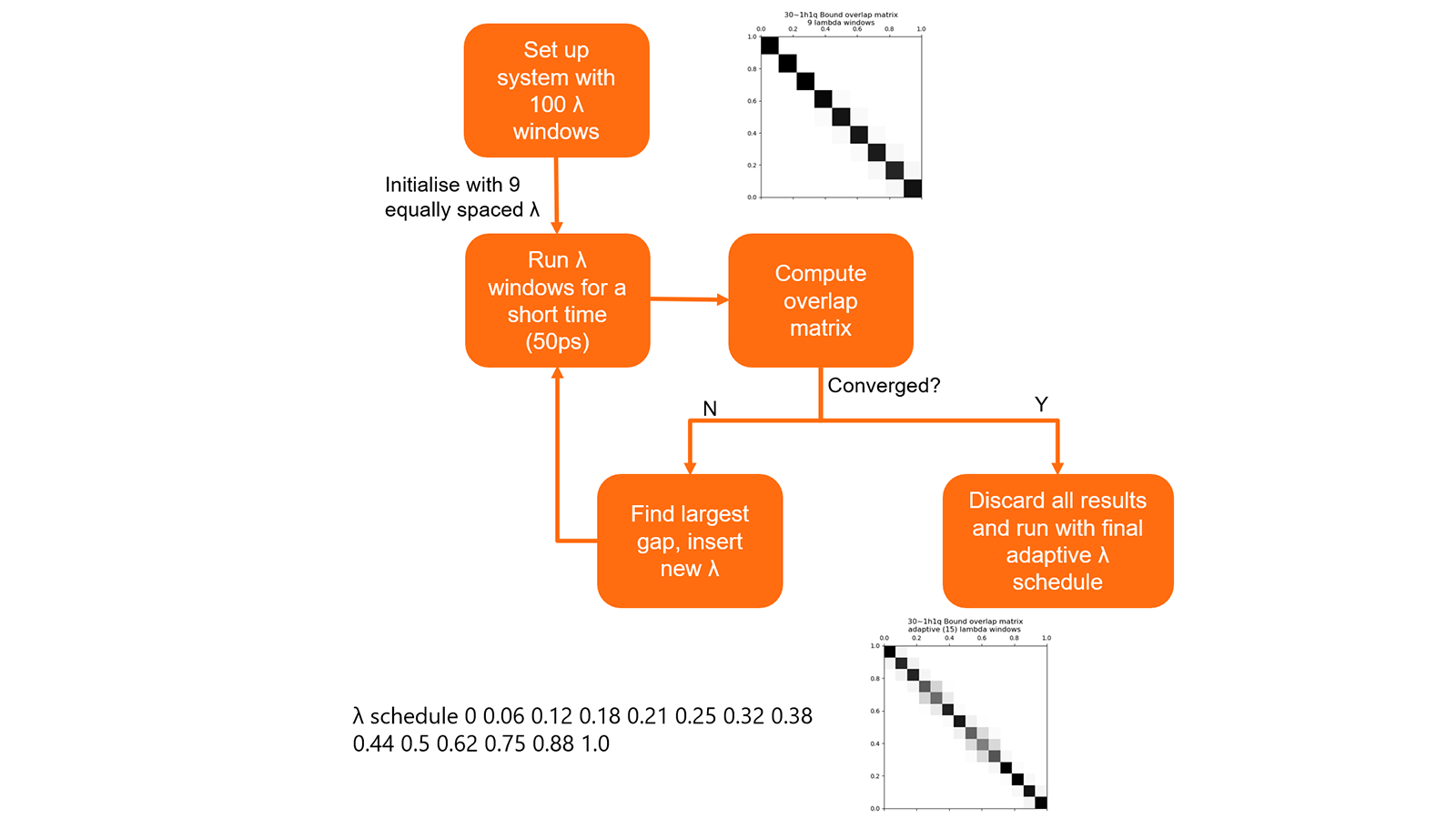
图8. 为链接规划最佳λ的算法总结
最后,在某些情况下,特定配体的最近邻居可能距离太远而无法进行有效计算。Flare FEP可以很好地处理配体之间多达8-9个原子的情况下工作良好,尽管如果添加或移除的原子是高度极性的,则该数量会减少。在近邻的差异大于此值的情况下,引入中间结构通常很有用,因为从两个保守链接中获得的 ΔΔG 估计值比从一个激进的链接中获得的 Δ Δ G估计值更佳。在之前版本的Flare中,用户需要手动设计和引入此类中间体。从 Flare V5 开始,添加了启发式算法来自动生成合理的中间结构(图 9)。
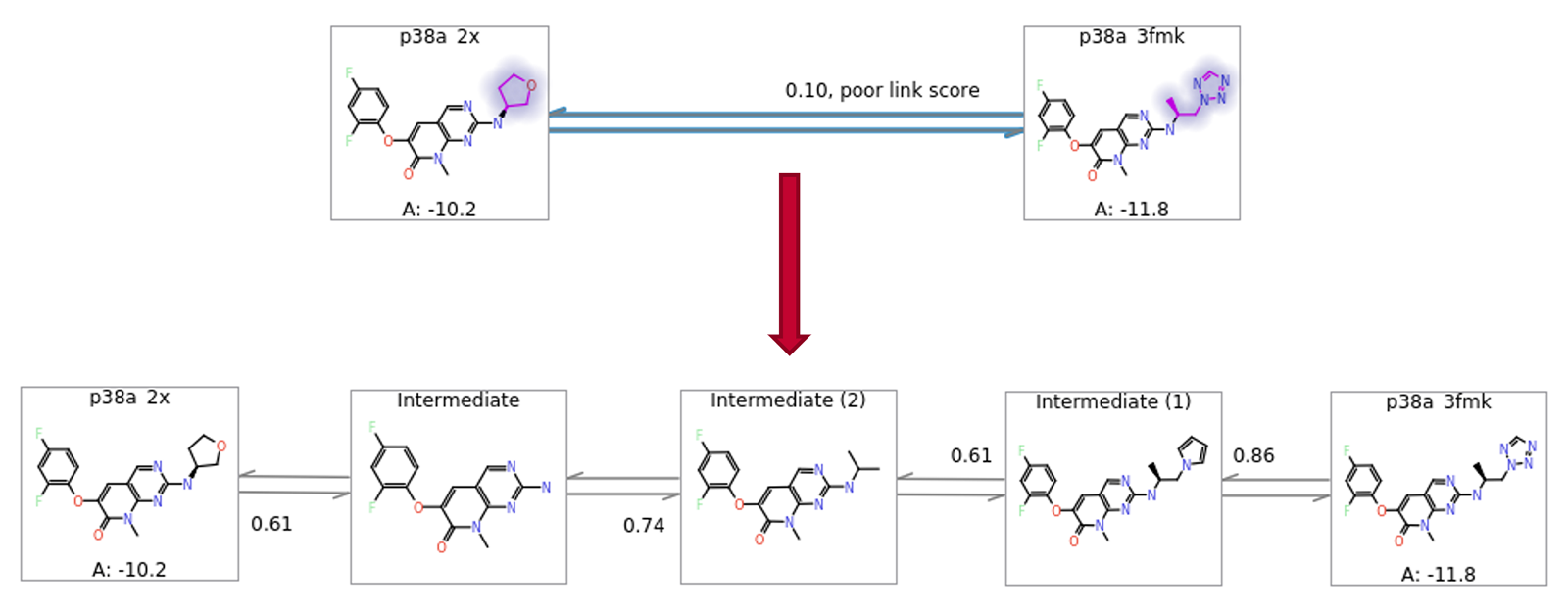
图9. 自动添加中间体结构以使得对大结构变化的ΔΔG评估更加健壮。
用Ignite™进行3D虚拟筛选
Ignite™是一种新技术,可在组合化学空间中实现快速3D虚拟筛选。大型数据库的常规虚拟筛选通常使用比较分子指纹来实现。这使得筛选速度相对较快,但缺点是打分最高的结果难以被实际命中。当然,分子在本质上是三维的,因此使用3D描述符可以得到更可靠的命中率。Ignite 3D描述符使用Cresset场技术[3]——该技术用给定构象的静电和形状来描述分子。
可合成的化学空间非常巨大,通常包含数十亿个分子[11]。这意味着完全枚举的库需要非常大的存储空间和计算资源来搜索。因此,一种更实用的方法是利用大多数化学空间的组合性质,即,将相对较少数量的砌块/片段组合起来以创建更多数量的枚举化合物。Enamine REAL Space就是这样的库,在撰写本文时,它拥有240亿种可能的化合物,这些化合物的可合成概率很高,约为80%。
Ignite算法通过在试剂空间中进行快速的初始筛选,解决了在如此大的空间中搜索的问题。每个虚拟库的试剂都被转换为“类产物”(product-like)的片段,将它们的反应官能团转换为库产物的最小表示。然后针对查询分子筛选这些产物样片段,以确定哪些片段可能具有显著的形状和静电相似性。
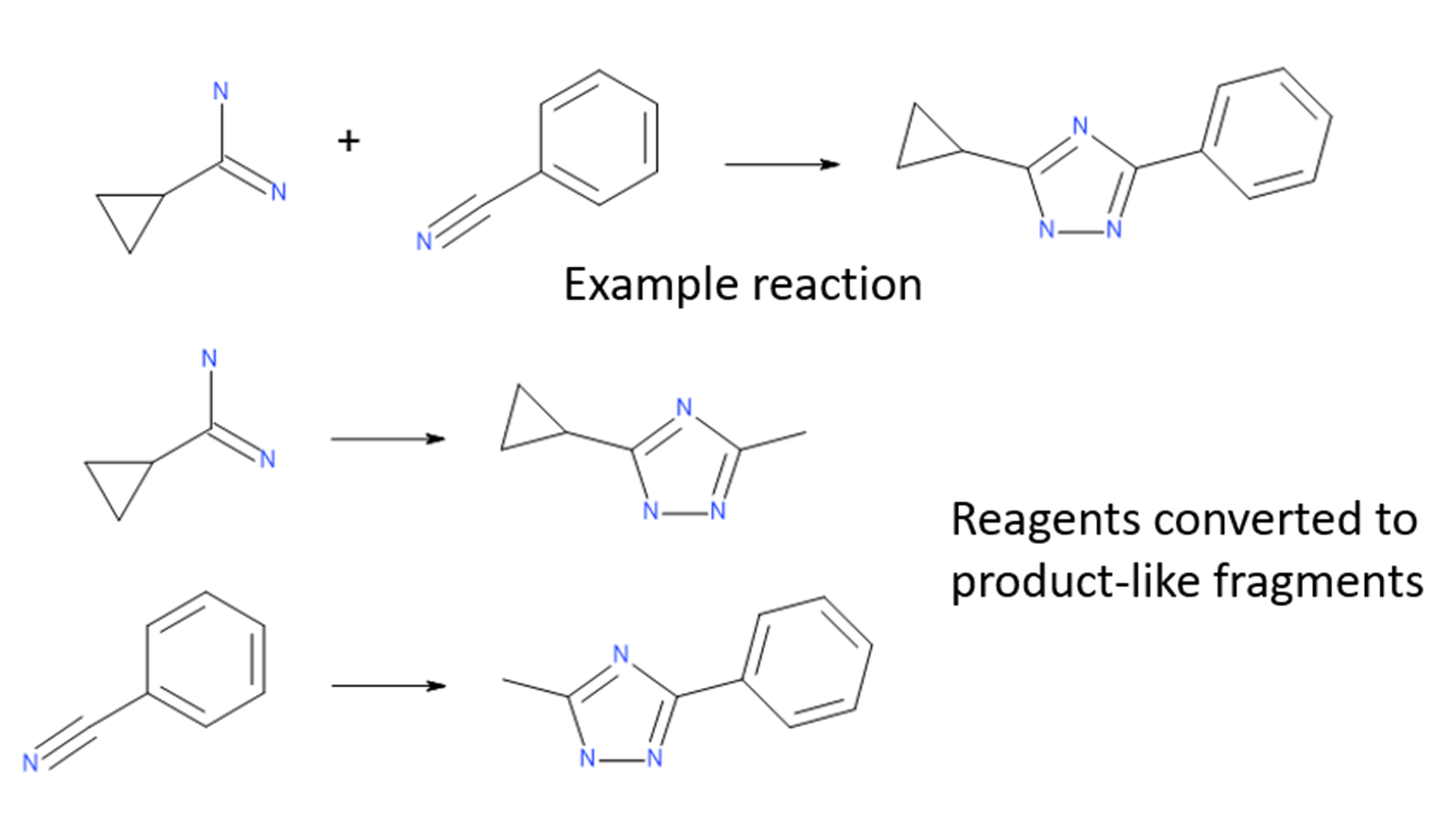
图10. 对于每个反应,将试剂库的试剂转化为产物样片段
只有得分最高的片段被传递到下一阶段,使用Spark™技术将之与来自试剂空间另一半的合成子组合成一个新分子。然后将组合分子与查询分子进行叠合并再次打分,该策略如图11所示。
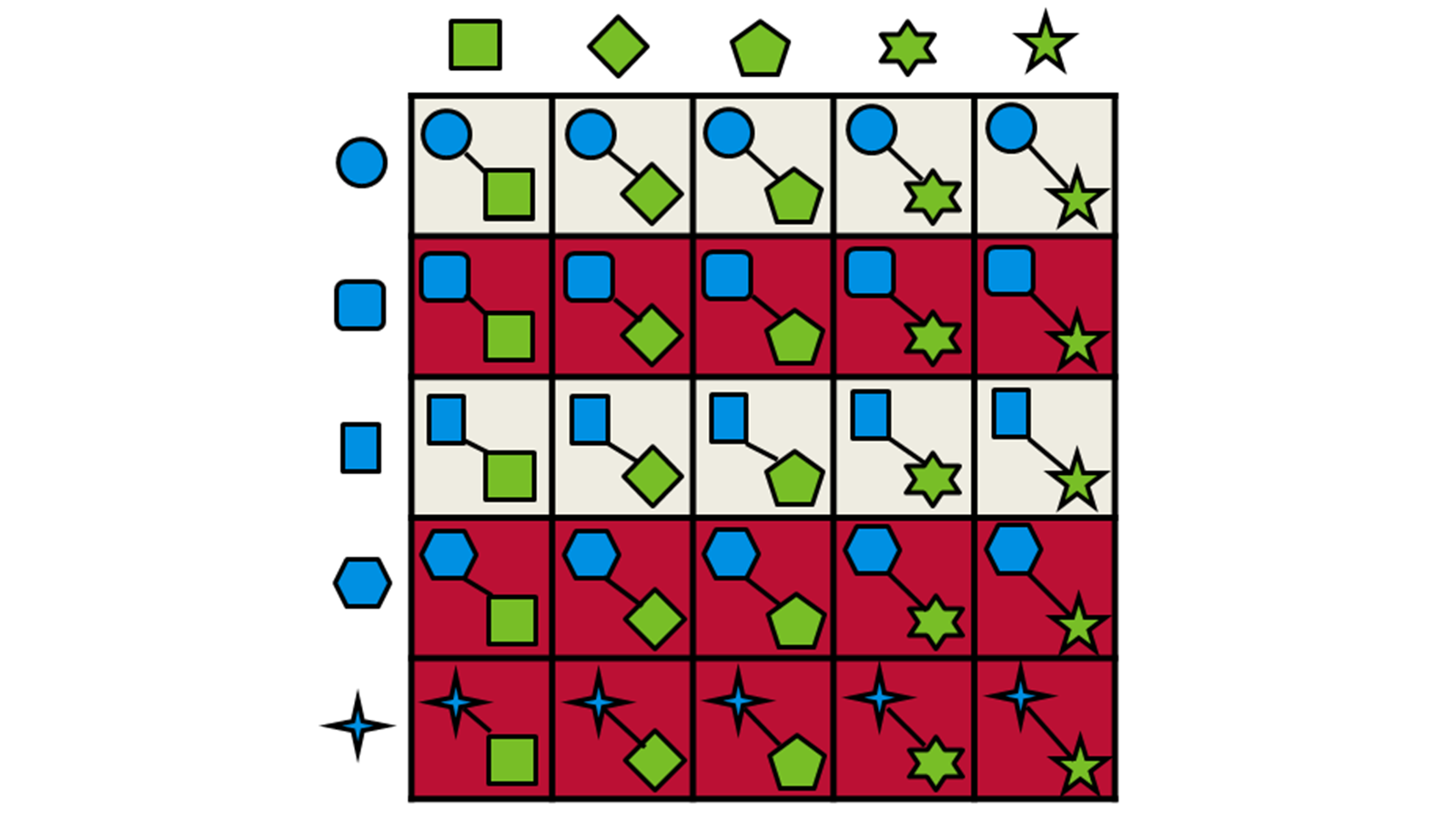
图11. Ignite使用的片段组合和产物选择过程的示意图。
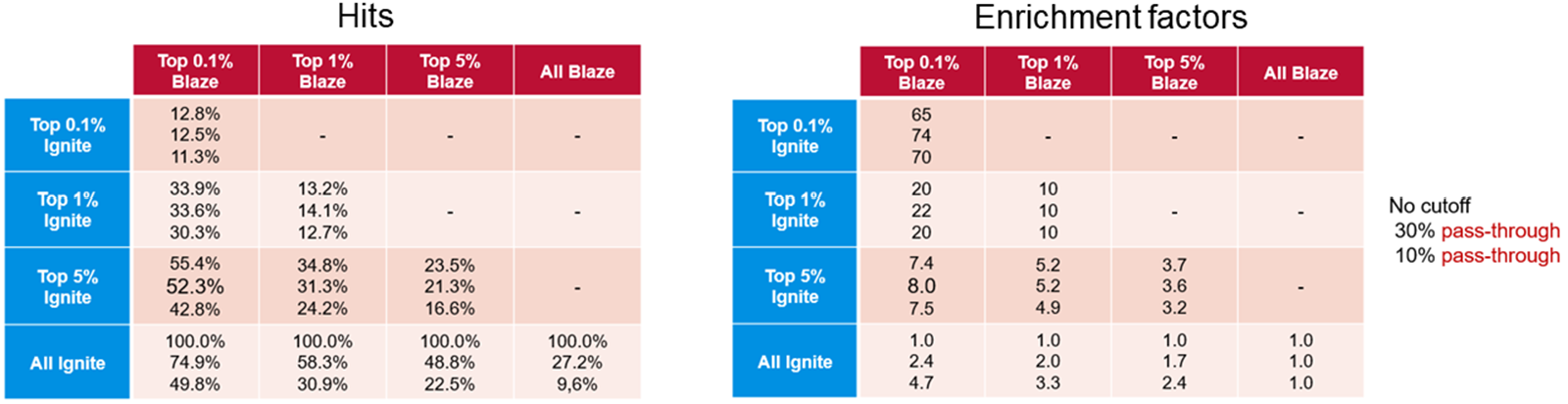
下表1比较了Ignite和Blaze™获得的富集因子。Blaze是一种基于配体的 3D虚拟筛选解决方案,可用于枚举空间的虚拟筛选,因此适用于数百万个分子的化合物库。在包含一百万个分子的验证库空间中,我们将使用Blaze检索到的结果与使用更快的Ignite方法获得的结果进行了比较。结果表明,Ignite可以检索高比例的Blaze命中化合物,而仅需筛选总化学空间的一小部分。将Ignite应用于更大的空间会产生更好的性能:在我们的验证合作伙伴Bayer的手中, Ignite可以在100个CPU上不到4小时的时间内完成整个Enamine化合物库(大于100亿分子)的筛选。
表1. 以Blaze结果作为基线的Ignite结果(左)和富集因子(右)
目前Ignite作为一种专有技术服务通过外包应用于您的项目。Cresset发现服务拥有一支计算化学家专家团队,随时准备帮助您加速商业资产的创建。联系我们进行免费的保密讨论,以进一步探索合作方案。
文献
- Hunter, C. A.; Sanders, J. K. M. The Nature of .Pi.-.Pi. Interactions. J. Am. Chem. Soc. 1990, 112 (14), 5525–5534. https://doi.org/10.1021/ja00170a016.
- Vinter, J. G. Extended Electron Distributions Applied to the Molecular Mechanics of Some Intermolecular Interactions. II. Organic Complexes. J. Comput. Aided. Mol. Des. 1996, 10 (5), 417–426. https://doi.org/10.1007/BF00124473.
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46 (2), 665–676. https://doi.org/10.1021/ci050357s.
- E. L. Mehler, The Lorentz-Debye-Sack theory and dielectric screening of electrostatic effects in proteins and nucleic acids, in Molecular Electrostatic Potentials: Concepts and Applications, Theoretical and Computational Chemistry Vol. 3, 1996
- Cheeseright, T. J.; Mackey, M. D.; Melville, J. L.; Vinter, J. G. FieldScreen: Virtual Screening Using Molecular Fields. Application to the DUD Data Set. J. Chem. Inf. Model. 2008, 48 (11), 2108–2117. https://doi.org/10.1021/ci800110p.
- Bauer, M. R.; Mackey, M. D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein-Ligand Complexes. J. Med. Chem. 2019, acs.jmedchem.8b01925. https://doi.org/10.1021/acs.jmedchem.8b01925.
- Xu, D.; Li, H.; Zhang, Y. Protein Depth Calculation and the Use for Improving Accuracy of Protein Fold Recognition. J Comput Biol 2013, 20 (10), 805–816. https://doi.org/10.1089/cmb.2013.0071.
- Xu, D.; Zhang, Y. Generating Triangulated Macromolecular Surfaces by Euclidean Distance Transform. PLoS ONE 2009, 4 (12), e8140. https://doi.org/10.1371/journal.pone.0008140.
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M. K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137 (7), 2695–2703. https://doi.org/10.1021/ja512751q.
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A. S. J. S.; Mackey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60 (6), 3120–3130. https://doi.org/10.1021/acs.jcim.0c00165.
- Warr, W. A.; Nicklaus, M. C.; Nicolaou, C. A.; Rarey, M.Exploration of Ultralarge Compound Collections for Drug Discovery, J. Chem. Inf. Model. 2022, 62, 9, 2021-2034. https://doi.org/10.1021/acs.jcim.2c00224
Cresset药物设计软件
Cresset杰出的分子设计软件
创新科技,直观软件