Cresset的CADD工作台Flare第8版带来了新的和增强的科学功能和方法,包括MM/GBSA方法用于计算配体-蛋白质复合物结合自由能,显著扩展了Flare FEP应用范围,用于GIST水分析的大正则非平衡候选蒙特卡罗(GCNCMC),QM进行HOMO/LUMO轨道的计算和显示,组合库枚举新增100多种反应,蛋白质和同源模建新增功能。
在此版本中,我们还进一步扩展了高度可视化的工具选择,以排除Flare FEP实验的故障并分析动力学轨迹。为分子动力学模拟和FEP模拟创建配体自定义参数以及为进一步研究准备配体结构的工具也得到了进一步开发和增强。
此外,可以应用新的Flare配置文件,使Flare GUI与当前的许可级别保持一致,从而使平台更高效、更易于使用。
用MM/GBSA单点计算对化合物进行优先级排序
MM/GBSA1是一种计算配体-蛋白质复合物结合自由能的方法,在准确性和计算效率之间取得了很好的平衡。它在理论上比分子对接等经验打分函数更严格,在各种体系中提供了更准确的结果(图1)。同时,它的计算成本低于Flare FEP等相对结合自由能模拟,使其可以直接从Flare GUI快速地对几百个化合物进行打分,或从命令行快速地对几千个化合物打分。
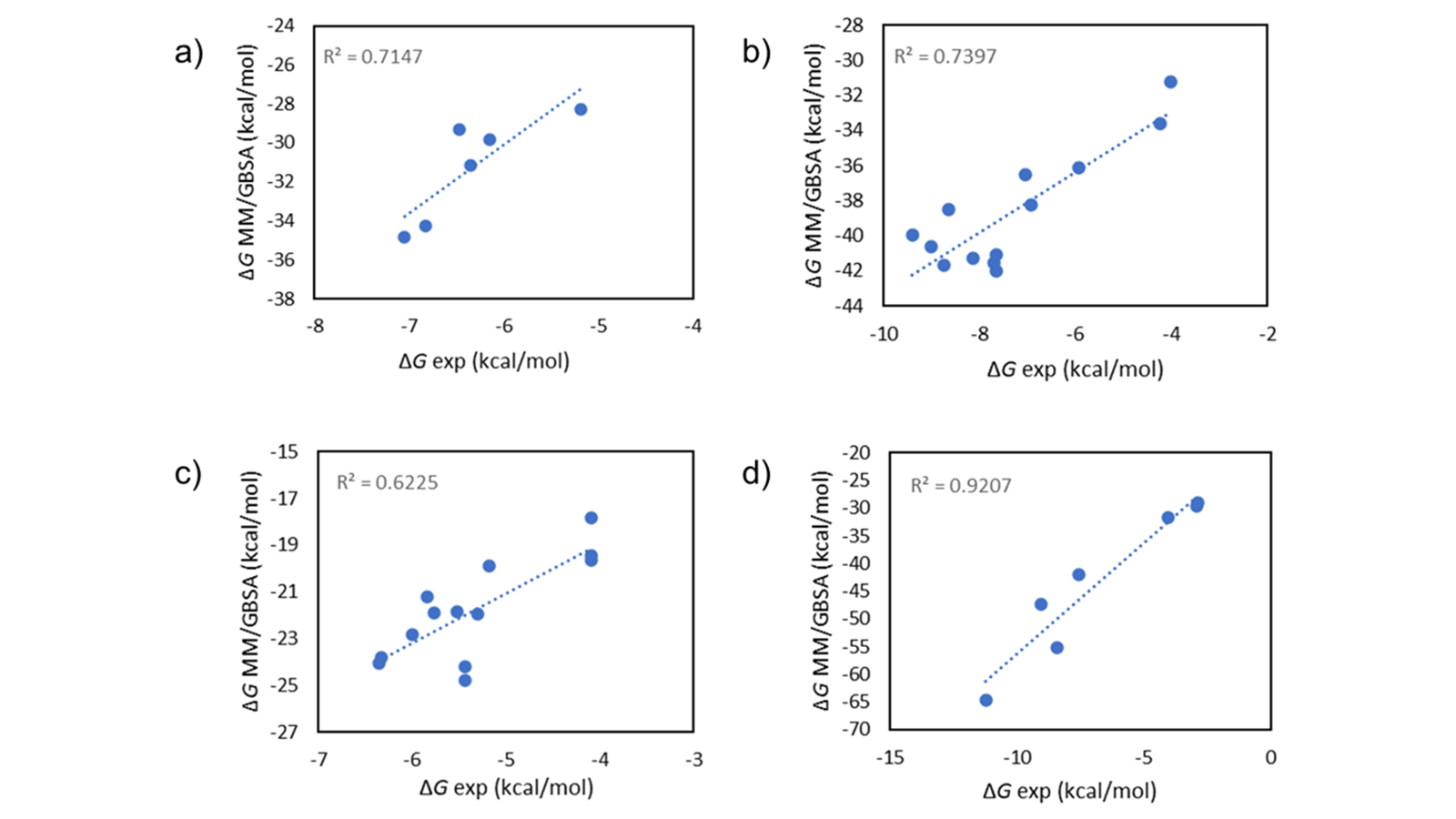
图1. 在FBDD数据集2上用Flare MM/GBSA单点打分的实验vs计算∆G值。
因此,MM/GBSA可用于高通量评估同系物配体的结合亲和力,例如在Flare Docking或Spark™ R-group替换实验后,使您能够自信地对您设计的分子进行优先级排序。
Flare MM/GBSA可以灵活使用(图2),可以选择使用不同版本的OpenFF以及AMBER GAFF/GAFF2力场来参数化配体。有多种不同的隐式溶剂模型可供选择,使MM/GBSA能够适应您感兴趣的蛋白质-配体体系,同时可以选择最小化配体和蛋白质以减少弹性应变和蛋白质-配体体系内的潜在静电冲突,从而提高结合自由能预测的准确性。
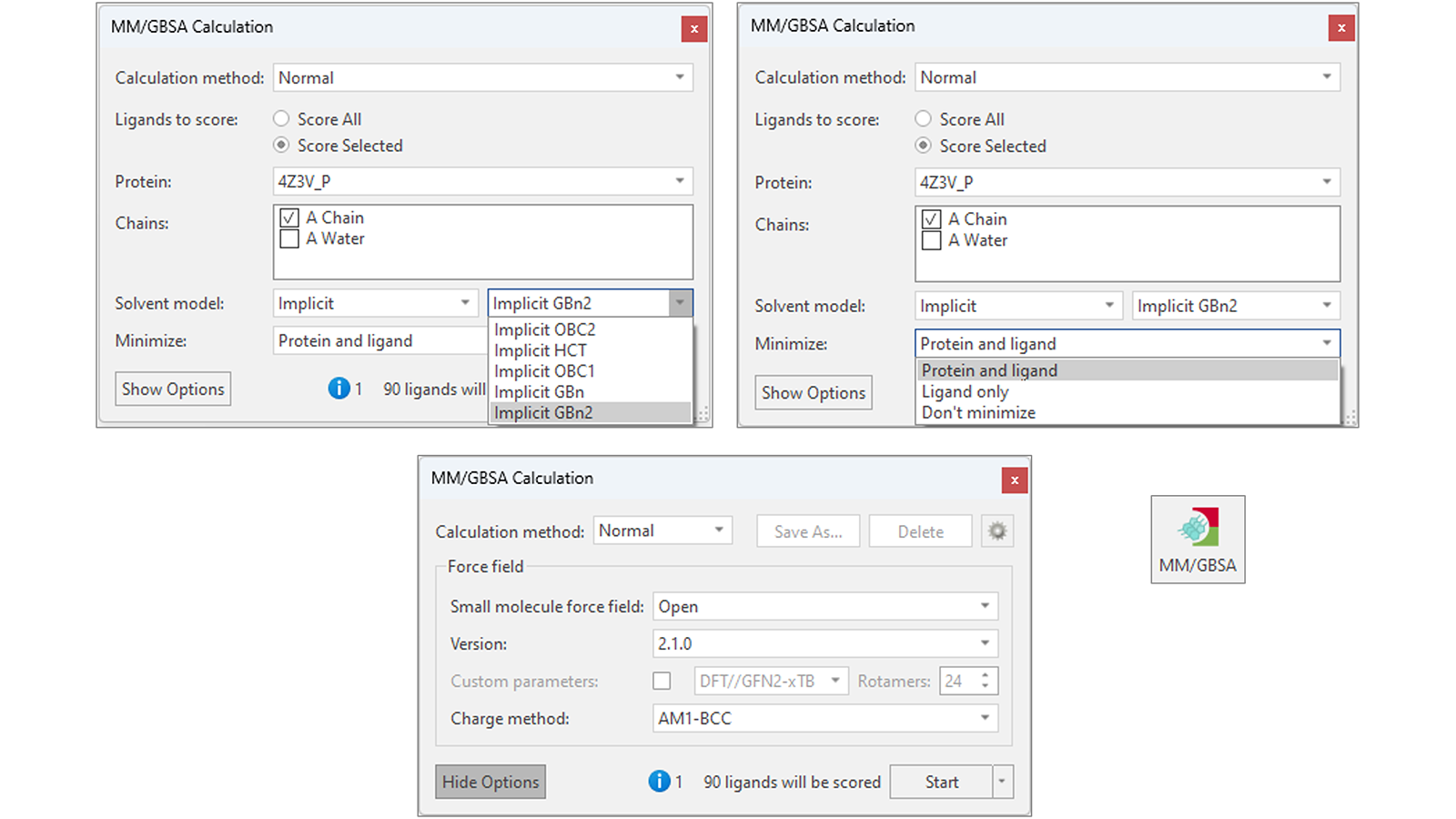
图2. Flare MM/GBSA提供几种不同的隐式溶剂模型(左上),可选对配体与靶标蛋白进行最小化(右上),可选不同的小分子力场对等待打分的配体进行参数化(下)。
显著地扩大了Flare FEP的适用范围、提供了更多的分析工具
Flare FEP在这个新版本的Flare中支持包括具有不同净电荷的配体在内的项目,显著扩展了Flare FEP在更多现实药物发现项目中的适用性。对带电荷微扰网络的预测性能与文献中报告的数据一致:图3中显示了一个例子。
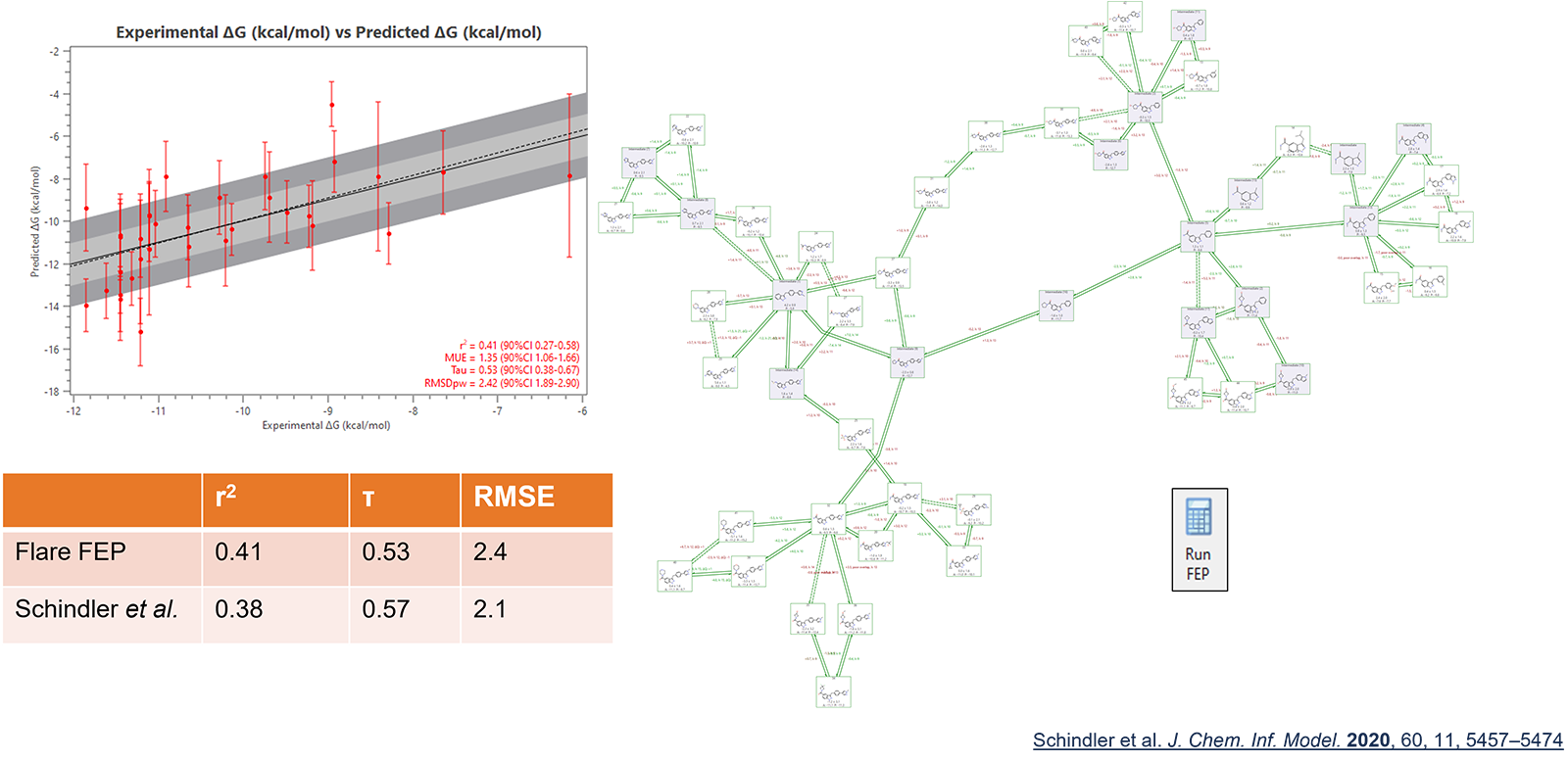
图3. Flare FEP在CDK8数据集上的预测性能与Schindler等人3文章报道的相比。
此版本还配备了其他高度可视化的工具,用于理解和排除Flare FEP计算结果中的问题。
扭转角分析图(图4)显示了参与特定Flare FEP转化配体的每个可旋转键的扭转角直方图,是在自由态和结合态的转化每帧中计算得到。该图可用于可视化起点和终点分子之间可能发生的意想不到的构象变化:这可能是体系采样不足的标志,可能需要重新运行更长时间的模拟。
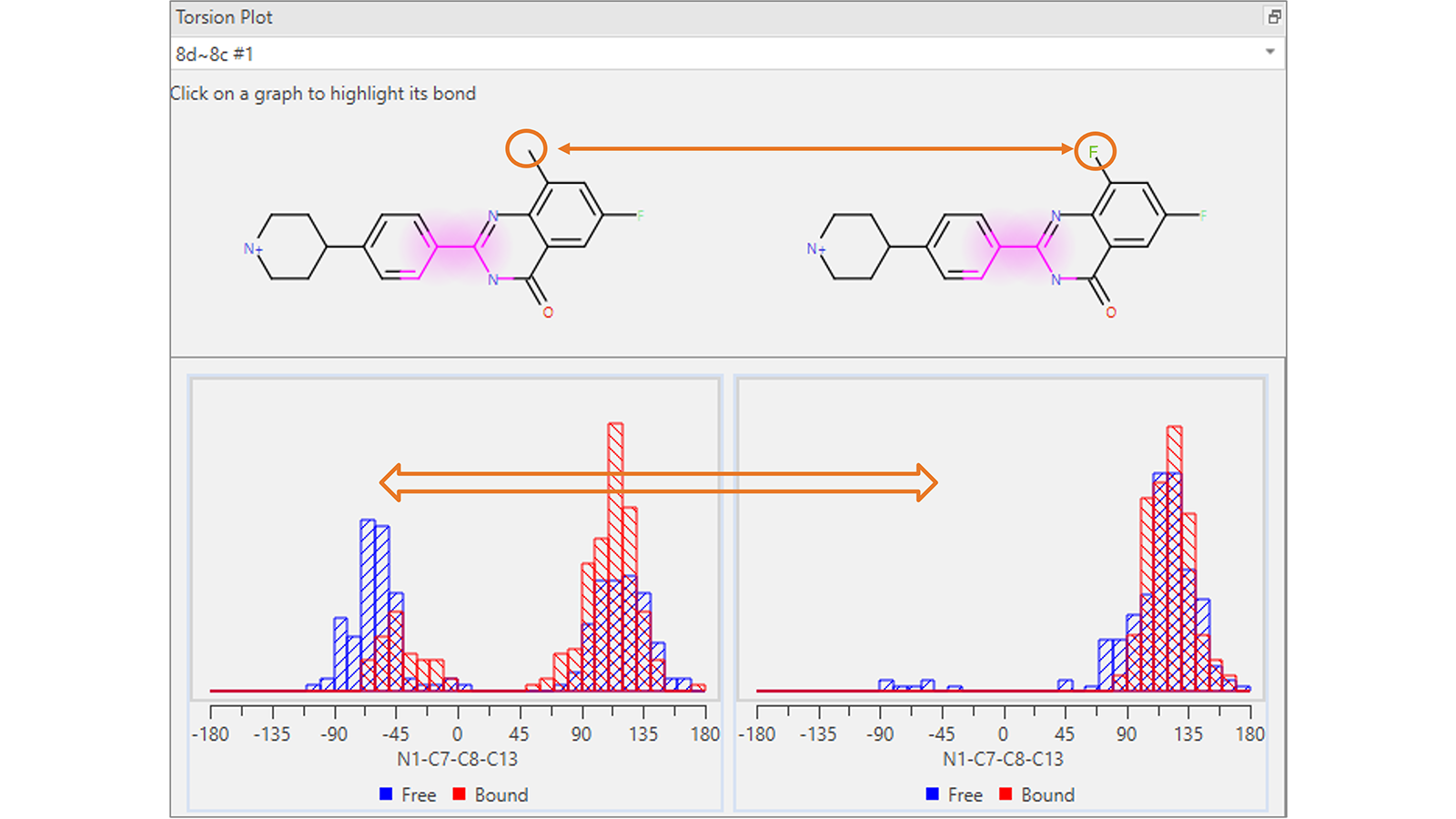
图4. 该扭转角分析图显示,在参与Flare FEP转换的分子公共骨架中,高亮显示的可旋转键的扭转角分布存在意想不到的差异。
收敛图可用于评估Flare FEP项目中每个转换的FEP模拟随时间的可靠性和收敛性。通过绘制模拟时间(正向线)的每个部分和轨迹(反向线)的相应最后部分的∆G和相关误差,它提供了一种非常有效的可视化工具来识别具有次优收敛性的转换,如图5所示的算例。在最佳收敛的转换中,正向线和反向线在图的右侧之前很好地相互接近:在图5中显示的转换没有发生这种情况,这可能需要更长时间的模拟。
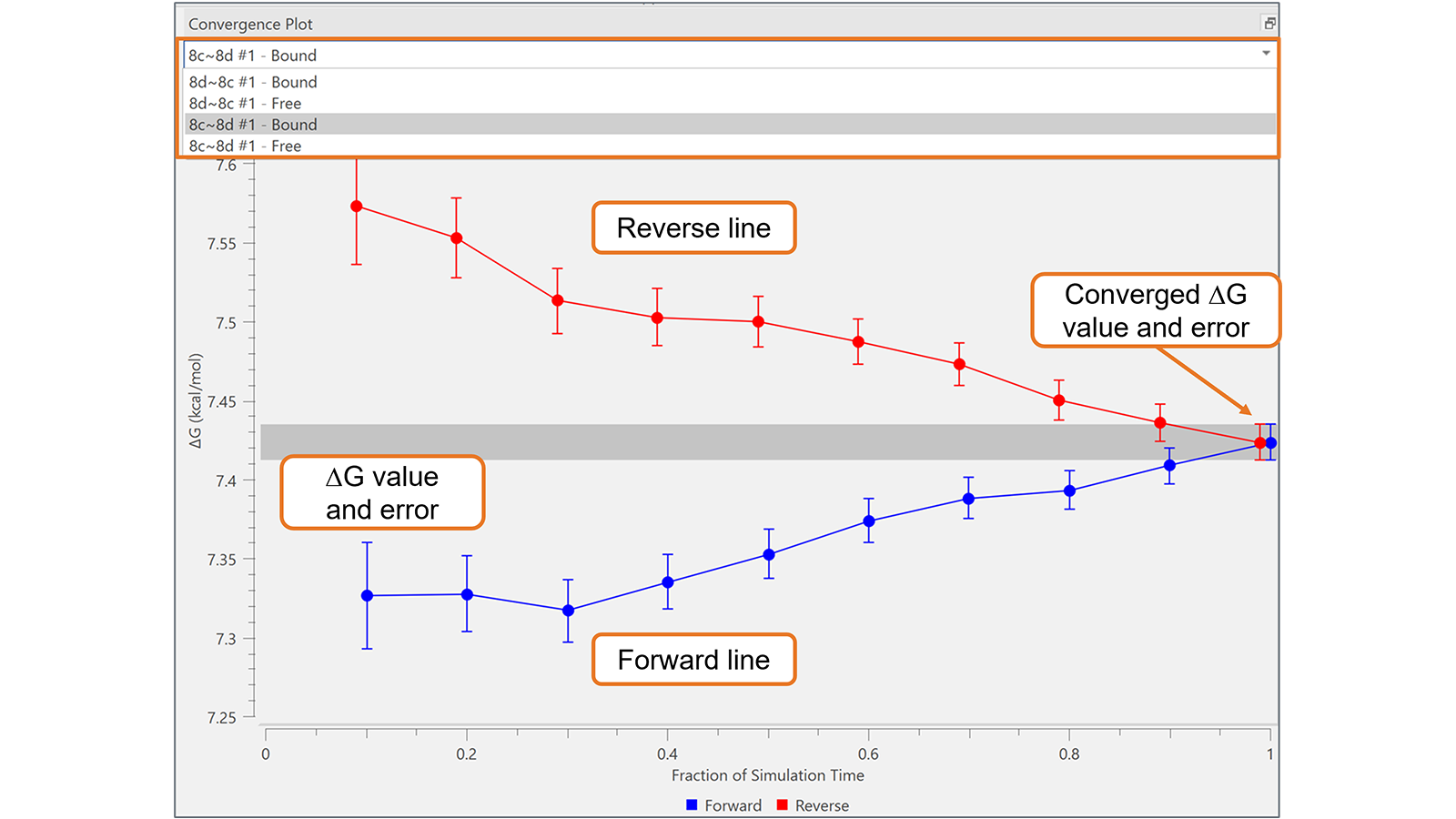
图5. 收敛图对于监控次优收敛的转化非常有用,如本例所示,这可能需要更长的模拟时间。
最后,新增的相互作用表单(图6)对于监测在转化的每个λ窗口期间被转化的配体与其相邻分子(蛋白质、水分子、辅因子等)之间形成的相互作用非常有用。高亮显示地意外改变的相互作用,使得识别值得注意的相互作用行为更加容易。
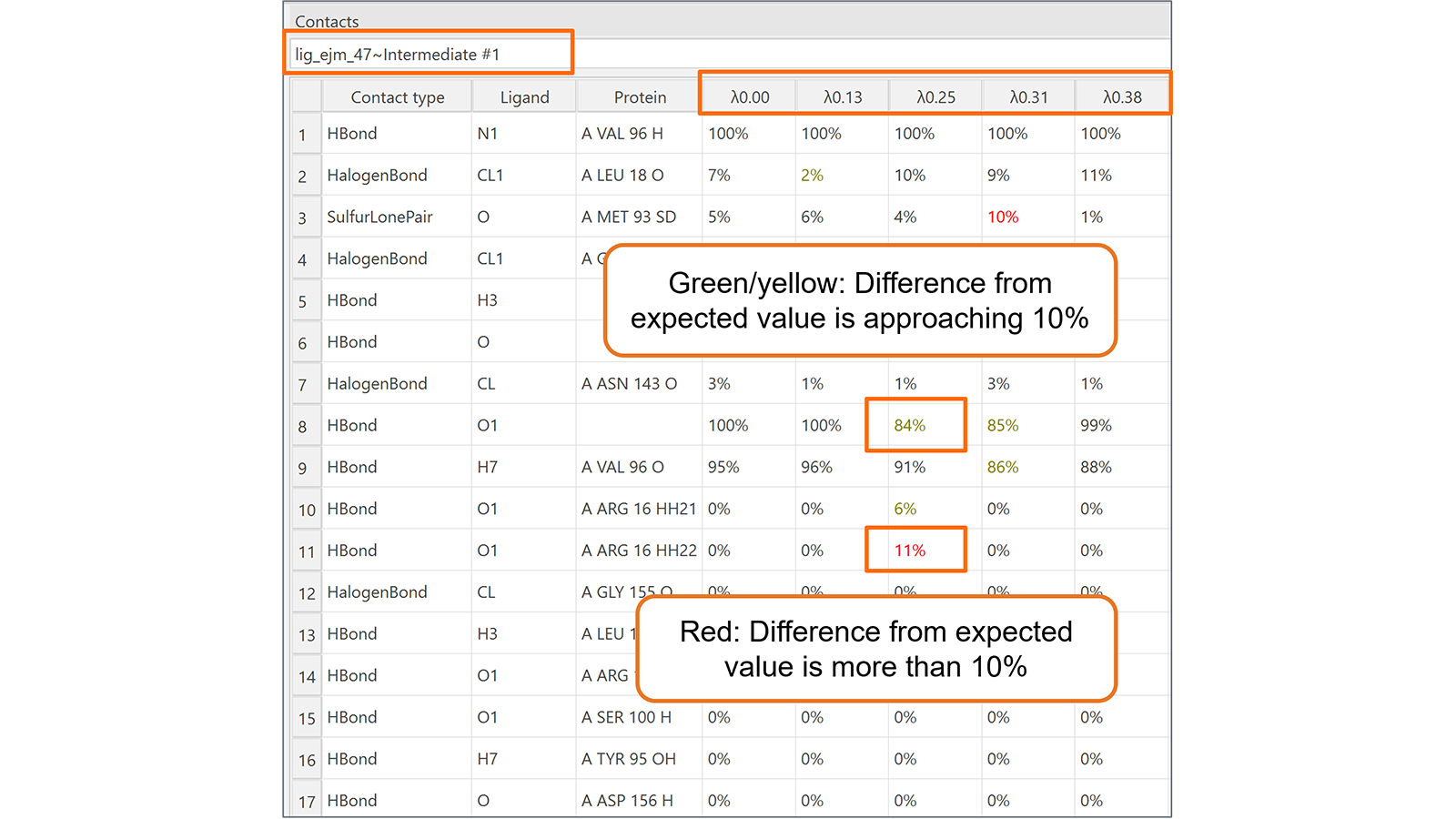
图6. 相互作用表单可用于监控Flare FEP计算在每个转换的lambda窗口期间配体与其相邻分子(蛋白质、水分子、辅因子等)之间形成的相互作用。
可视化配体的HOMO和LUMO轨道,深入了解配体的反应性
我们进一步扩大了Flare中针对小分子量子力学计算的实现,包括计算和显示HOMO和LUMO轨道的能力。这与HOMO/LUMO能隙(HOMO/LUMO gap)的计算一起非常有用,可以深入了解小分子的反应性和代谢行为。
例如(图7-顶部),从文献4中得知,2,4-二氯-5-甲基嘧啶与哌啶的亲核取代反应优先发生在4位,而不是2位。2,4-二氯-5-甲基嘧啶的LUMO计算和可视化解释了实验行为。C2和C5没有相关的LUMO波瓣,而C4和C6都与最大的波瓣相关:因此,亲核取代反应在这些位置之一是有利的。
在第二个例子中(图7,底部),检查萘的HOMO可以解释为什么硝酸的亲电攻击发生在C1(而不是C2)位置,因为这与较大的HOMO波瓣有关,表明这个位置电子更多。
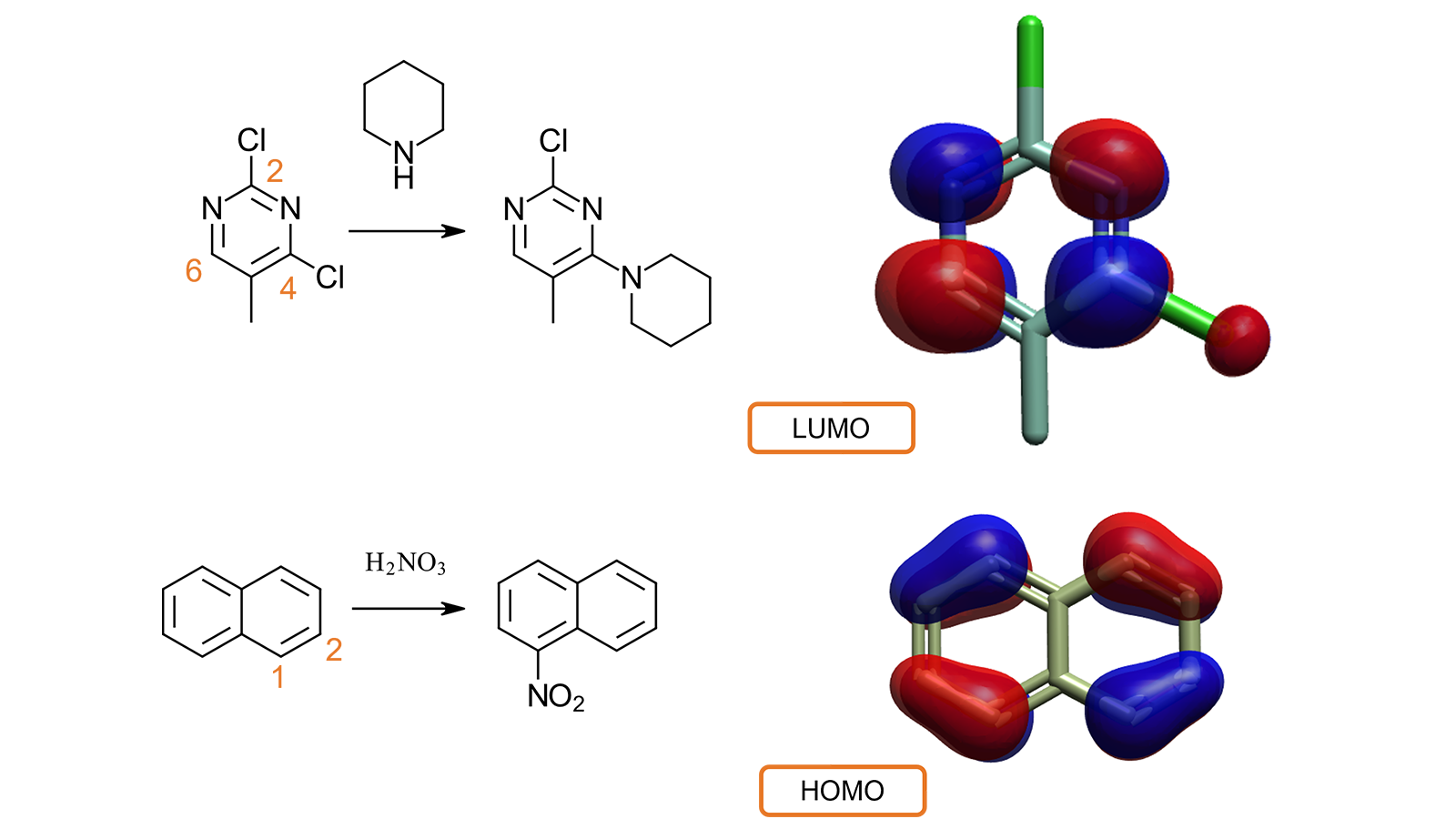
图7. 可视化LUMO与HOMO轨道有助于理解小分子的反应性与代谢行为
组合库枚举新增100多个真实的合成化学反应
我们大大扩展了Flare的预制枚举反应集,现在有150多个反应。这些广泛使用的合成化学反应涵盖了多种转化,包括单/双芳香环和非芳香环的形成、C-C和N|O|S-C键的形成、脱保护、氧化和还原反应。
新增“Search reaction”功能来快速地找到想要的反应(图8)。
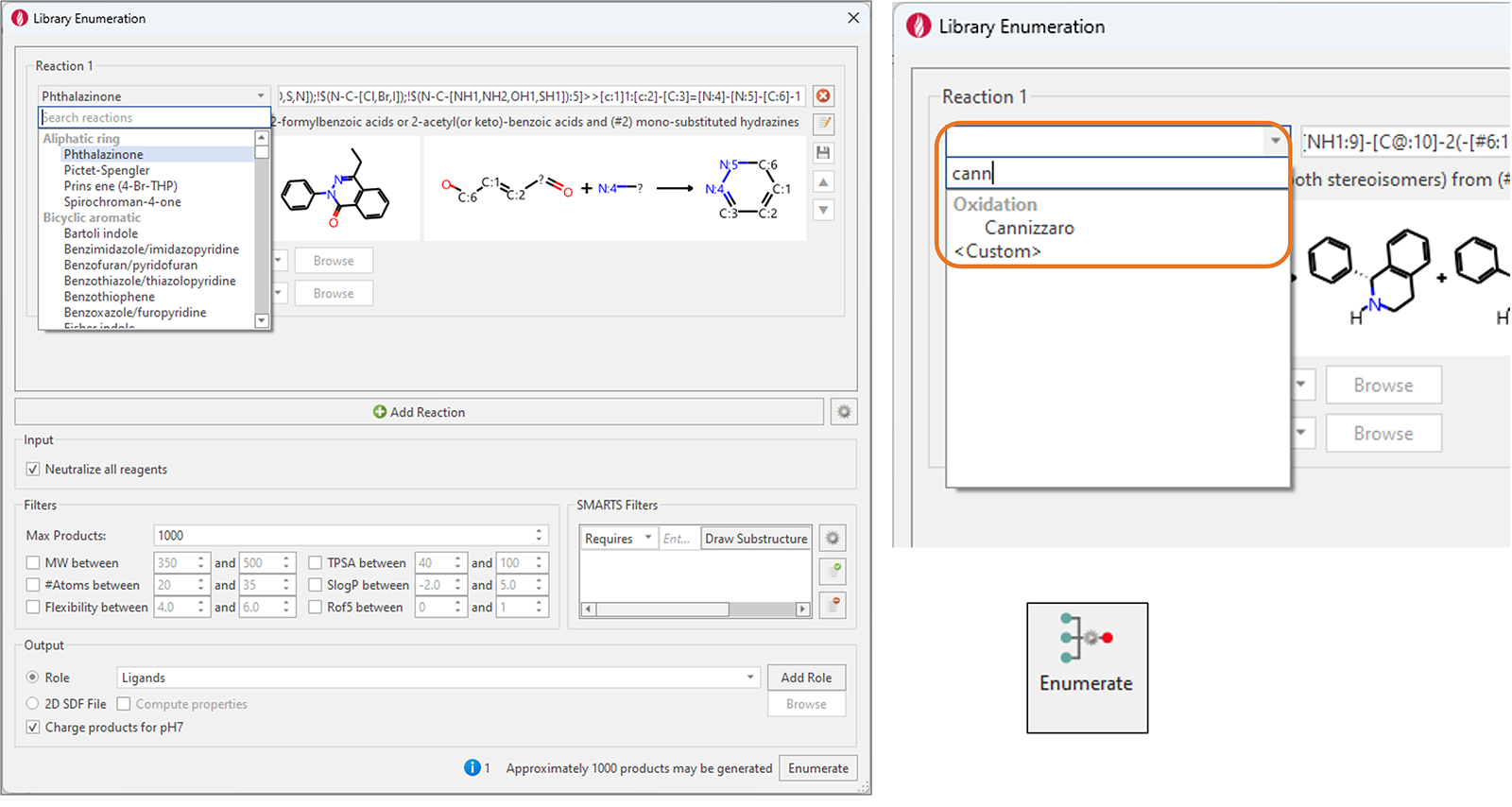
图8. 左:Flare V8组合库枚举功能新增100多个反应,涵盖了各种广泛使用的合成化学反应。右图:新增“Search reactions”功能使查找所需的反应变得容易。
综合性配体准备
健壮的配体准备是准备小分子以进一步分子模拟研究所必需的重要步骤,包括修复质子化状态、互变异构状态、脱盐以及枚举(必要时)未定义的立体中心。
在Flare V8中,我们新增“Pop to 3D and minimize”选项(图9),通过为您的配体生成合理的3D结构来完成配体准备,为进一步的计算做好准备,如对接和打分或基于配体的叠合。
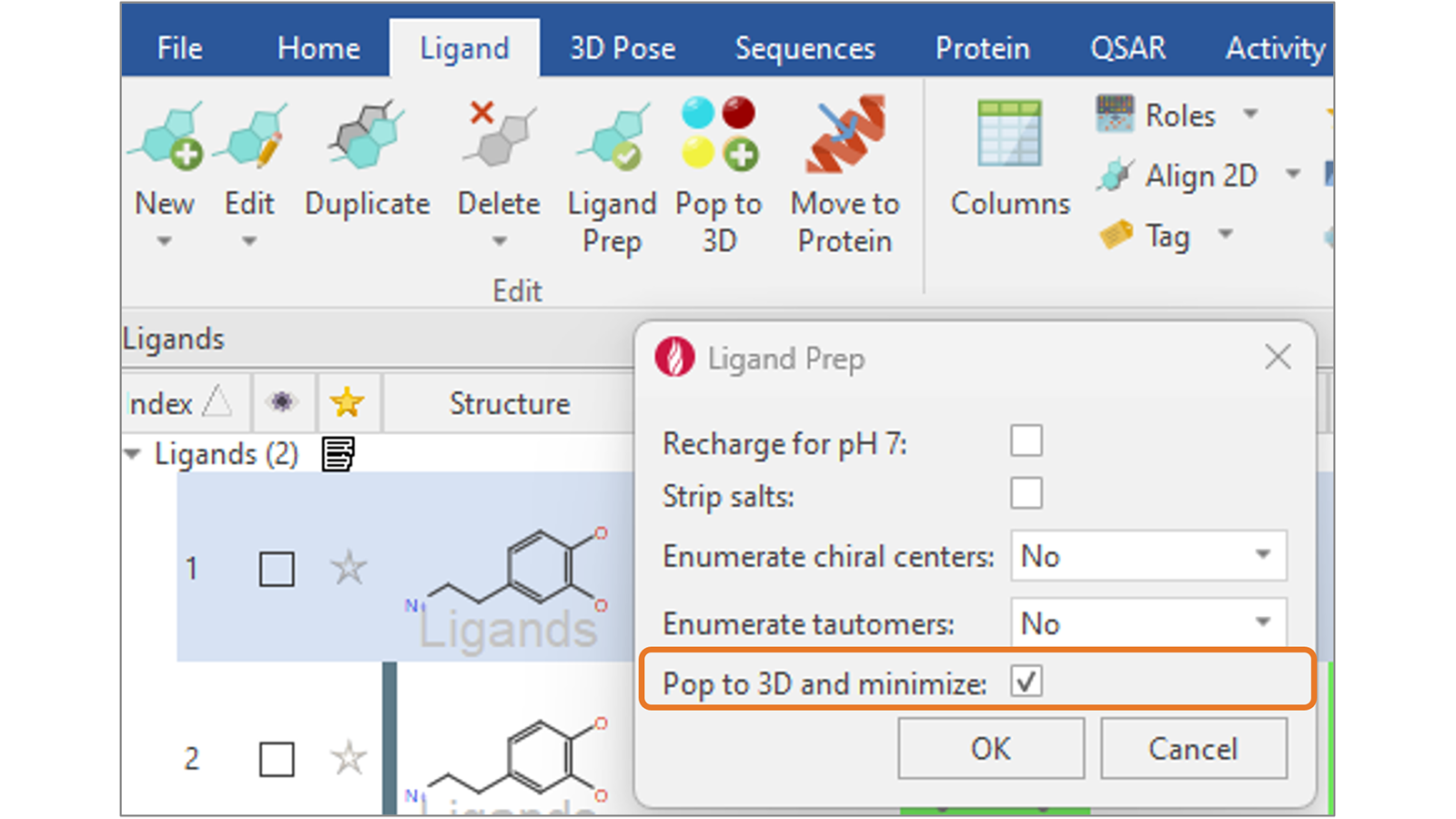
图9. 用新增的”Pop to 3D and minimize”选项完成配体准备,为你的配体生成有意义的3D结构
新增与增强的分子动力学模拟分析工具
Flare中高度可视化的工具集新增了“Radius of Gyration”(RG,回转半径)图,如图10所示,可以用来分析分子动力学模拟研究的结果。
该图显示了蛋白质α碳的RG值随时间的变化,可用于表征生物分子(通常是蛋白质或肽)的紧凑性或大小。较小的RG值表示更紧凑或紧密折叠的结构,而较大的RG值表示更伸展或无序的构象。因此,RG图可用于研究生物分子的结构变化、柔性和动力学,这对于理解它们的功能和行为至关重要。
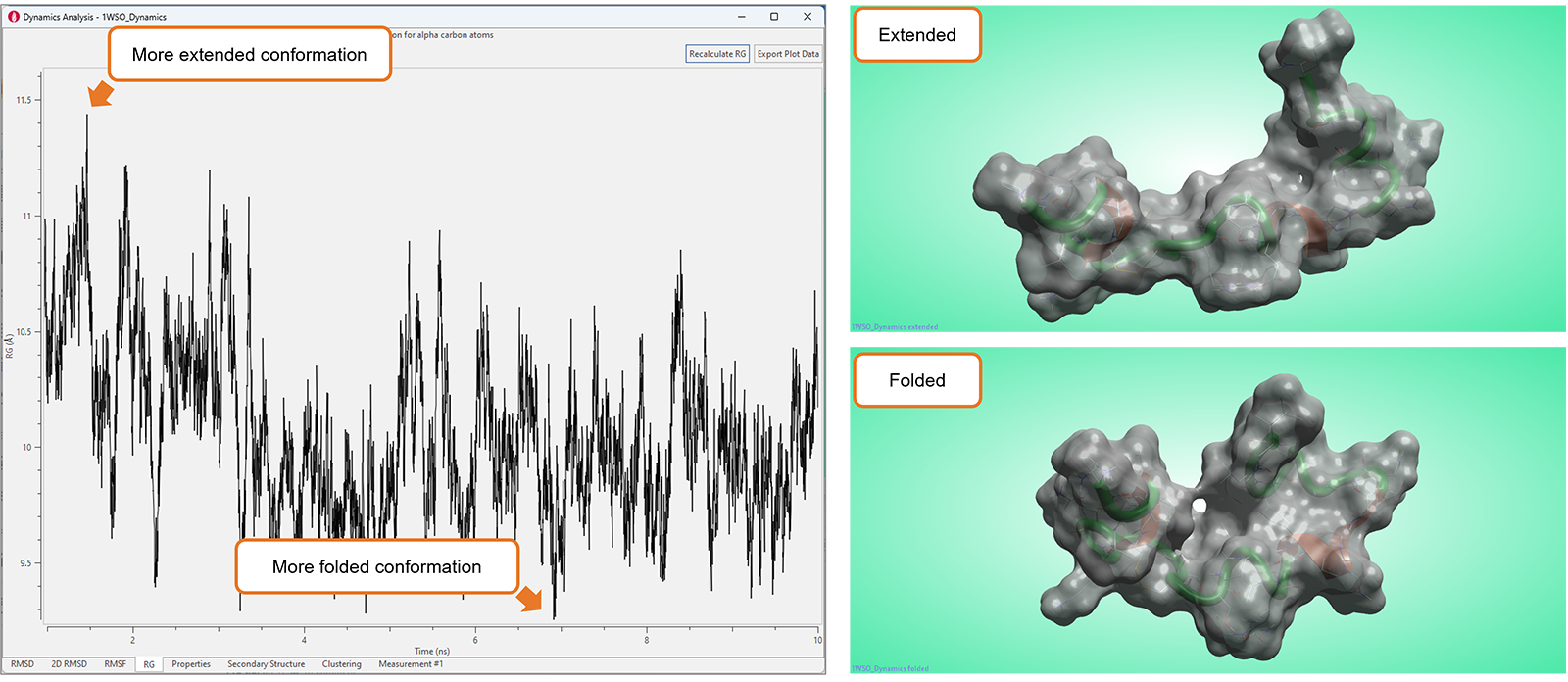
图10. 在RG图中显著的波动典型地对应于生物分子大的构象变化
在Flare V8中,我们还进一步增强了相互作用分析表单,报告模拟过程中观察到的有利配体-蛋白相互作用的统计数据,包括配体与水分子的相互作用(图11)。
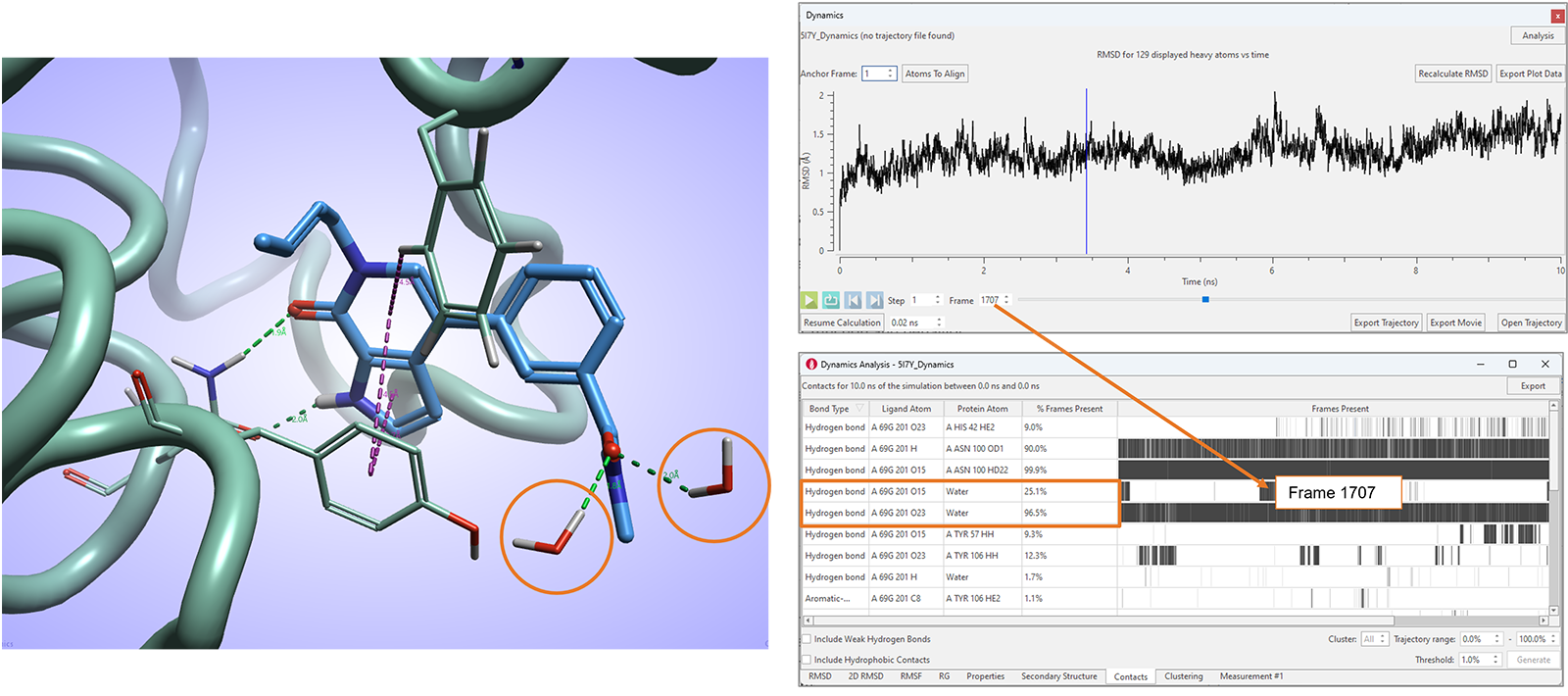
图11. Flare V8的相互作用表单对应有利的配体-蛋白与配体-水相互作用
Flare中分子动力学模拟的进一步增强包括了在无配体结合的apo活性位点上使用巨正则非平衡候选蒙特卡罗(GCNCMC)6,7的能力,以及导出动力学轨迹非连续帧的能力。
精确力场参数的生成与可视化以用于Dynamics与Flare FEP研究
通过专用的“Custom Parameters(自定义参数)”功能,为您的配体在Flare V8中生成精确的OpenFF扭转参数得到了简化。选择一个或多个配体后,按下此按钮会显示一个小部件,您可以选择OpenFF版本和生成自定义参数的方法(DFT//GFN2-xTB、GFN2-xTB或ANI-2X),以及用于计算的旋转异构体数量(图12右上)。
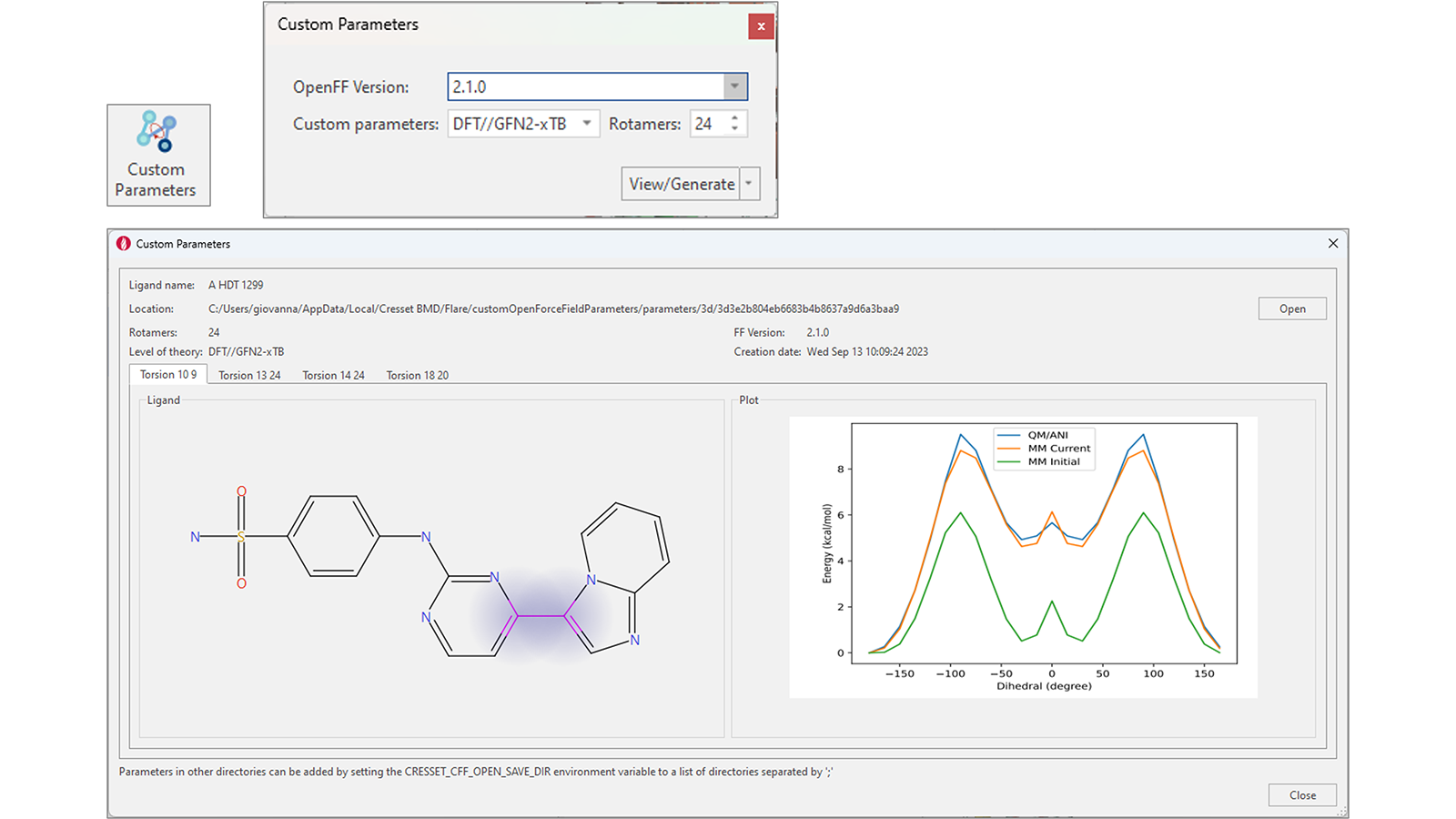
图12. 增强的工作流以用于生成精确的扭转角力场参数
在计算结束时,自定义参数可以存储在本地或共享位置,并将在后续的Dynamics和Flare FEP计算中自动使用。
如果自定义参数已经存在,则按下“View/Generate”按钮将打开一个窗口,显示感兴趣配体中每个可旋转键的计算结果(图12 – 底部)。
蛋白与同源建模新增工具
Flare的这一版本包括新的和增强的工具,以促进蛋白质结构的建模。
“Protein”选项卡中的“New Conformation”功能8可用于探索所选残基侧链可供选择的其它构象、合理的构象。您可以通过反复按下按钮一次探索一个侧链构象,或通过选择相应的选项创建所有构象,以避免与蛋白质其余部分发生空间冲突,这将把它们保存为备选构象(alternate conformations,图13)。
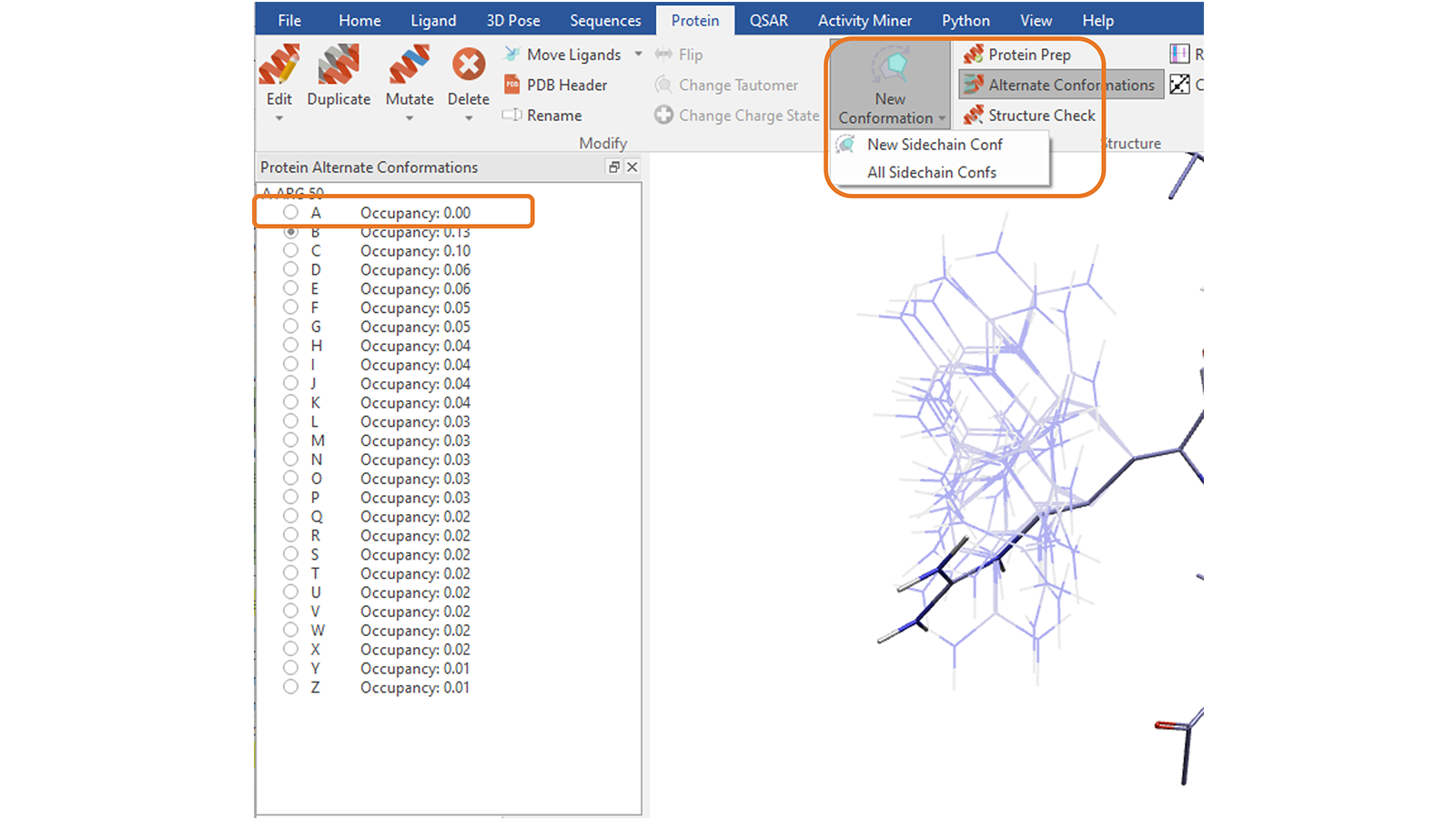
图13. 为挑选的蛋白质残基侧链一次创建一个构象或所有合理的构象。选择创建所有侧链构象,将把它们另存为备选构象,列表中的第一个条目是原始构象。
“Sequences”选项卡中添加了便于蛋白质和同源模建的新功能(图14),包括:
- FASTA与ClustalW文件格式的序列导入与导出
- 对选定的残基或整个链进行突变,以匹配另一个序列比对过的蛋白质链的序列
- 对选定的残基或整个链进行重新编号,选择是否对gap进行重新编号,并指定插入代码以避免重复编号
- 用于修复蛋白结构里的空隙(gap)的FREAD Loop Modeling方法移到此处以增强可用性
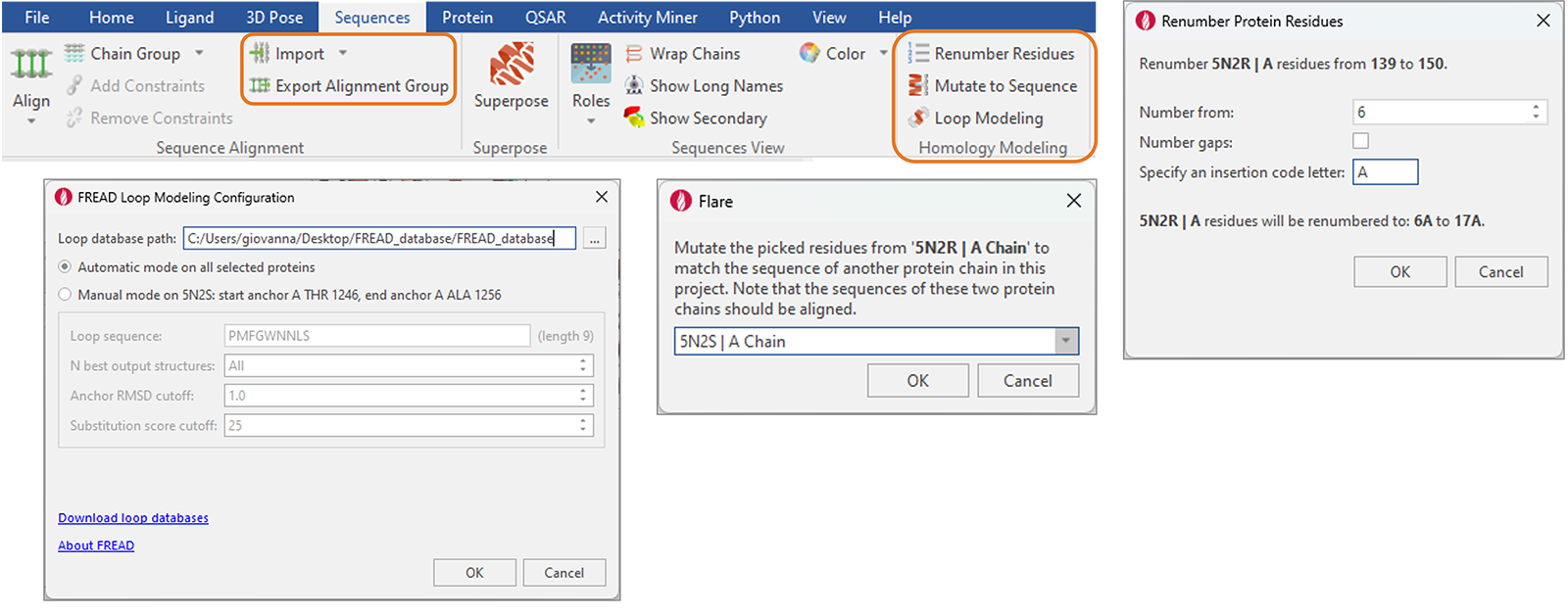
图14. Flare中重组的“Sequence”选项卡提供了蛋白质和同源模建的新增功能。
最后,比对表单有一个新的’Frequency Logo’可以直观地映射序列比对的一致性和多样性(图15):工具提示显示了在比对过的序列某个位置看到的所有残基及其频次。
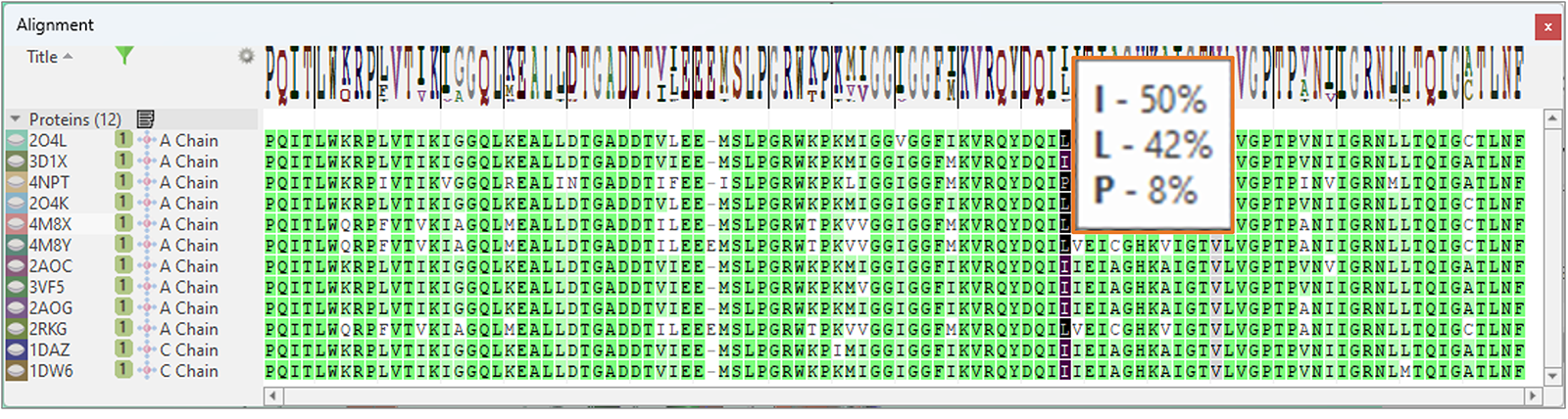
图15. ‘Frequency Logo’从视觉上与序列比对的一致性和多样性对应
增强的GIST水分析可以使用GCNCMC
本版本中的GIST水分析实验可以从增强的大正则非平衡候选蒙特卡罗(GCNCMC)的水采样6,7中受益,这对于确保蛋白质活性位点中隐匿口袋的最佳水合作用特别有用。
如图16所示,可以在计算的平衡步骤中,在以实验中包含的配体为中心的GCNCMC球体所定义的区域内,采取GCNCMC步骤。球体的大小和GCNCMC移动的频率可以在高级选项中进行微调。
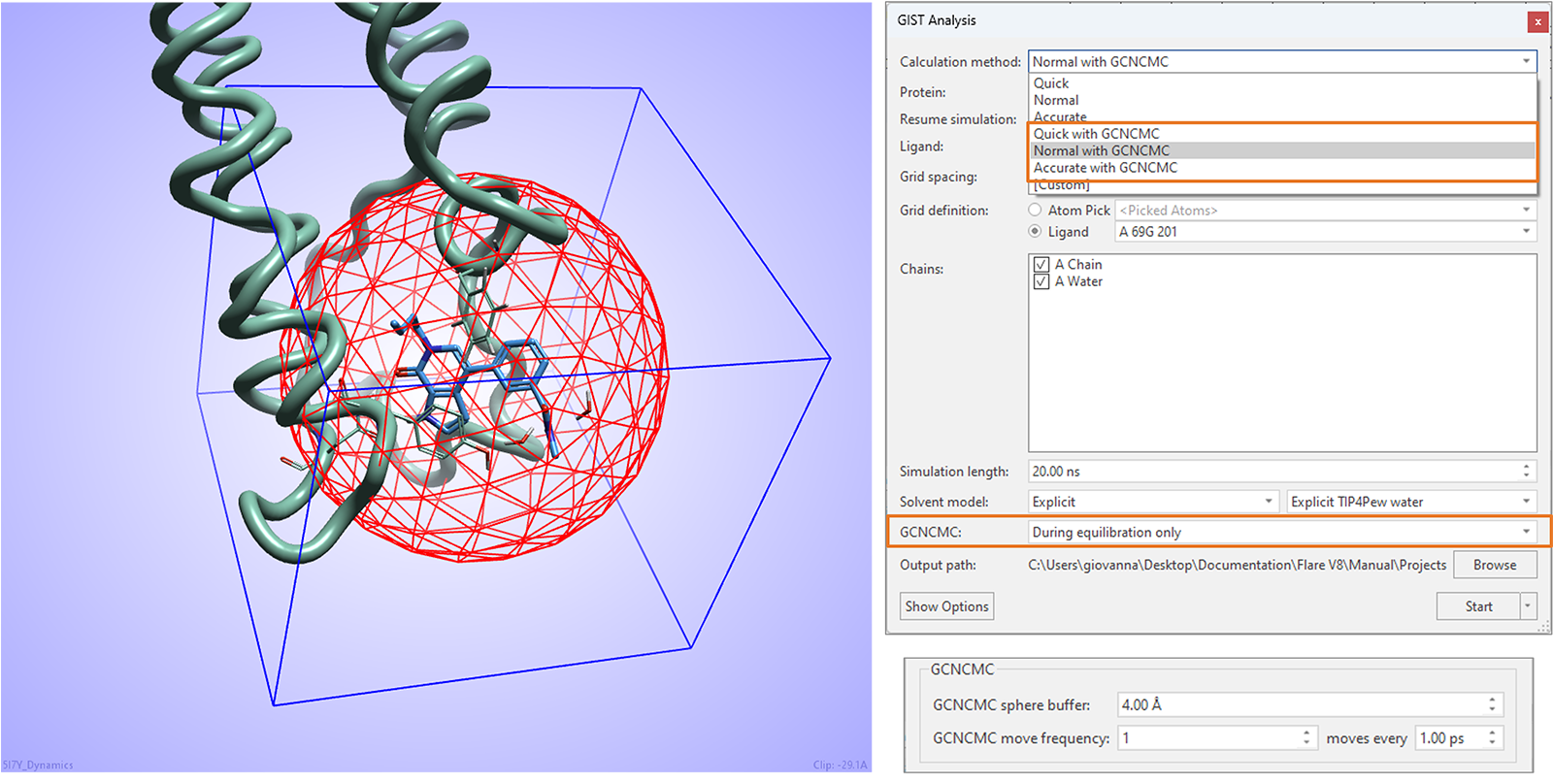
图16. Flare V8中的GIST水分析可以从实验平衡阶段采用GCNCMC中受益。
使用新的配置文件自定义Flare菜单的外观
Flare中的这个新功能可用于定制Flare功能区选项卡菜单,例如通过选择一个默认配置文件(图17)来匹配您当前的Flare许可证。通过这种方式,您当前许可证中不可用的选项卡和功能(按钮)将被隐藏:此外,所选配置文件将在您打开新项目或重新启动Flare时被记住。在任何时候,如果您想知道您可能错过了哪些令人兴奋的Flare功能,选择“完整”配置文件将带回所有Flare选项卡和按钮。
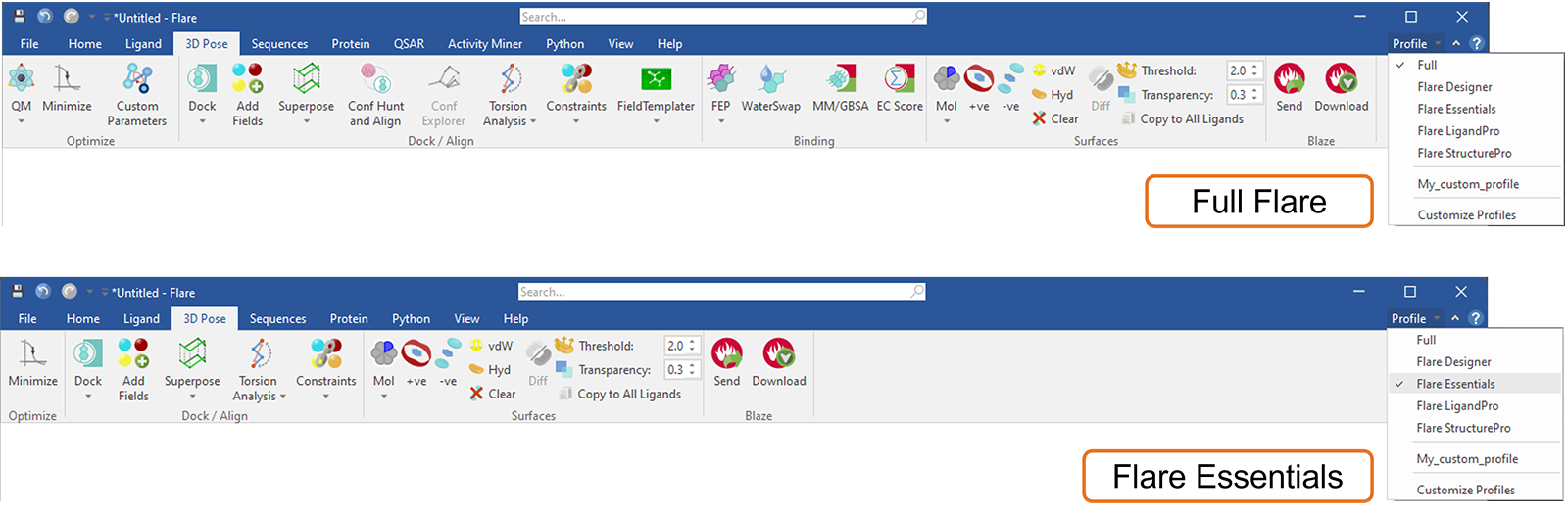
图17. 选择Flare Essentials配置文件来自定义Flare菜单,仅显示此许可级别包含的选项卡和按钮。
您还可以更进一步,创建一个自定义配置文件,仅显示您更频繁使用的选项卡和按钮,隐藏所有其他选项卡和按钮,并自定义它们的外观(图18)。
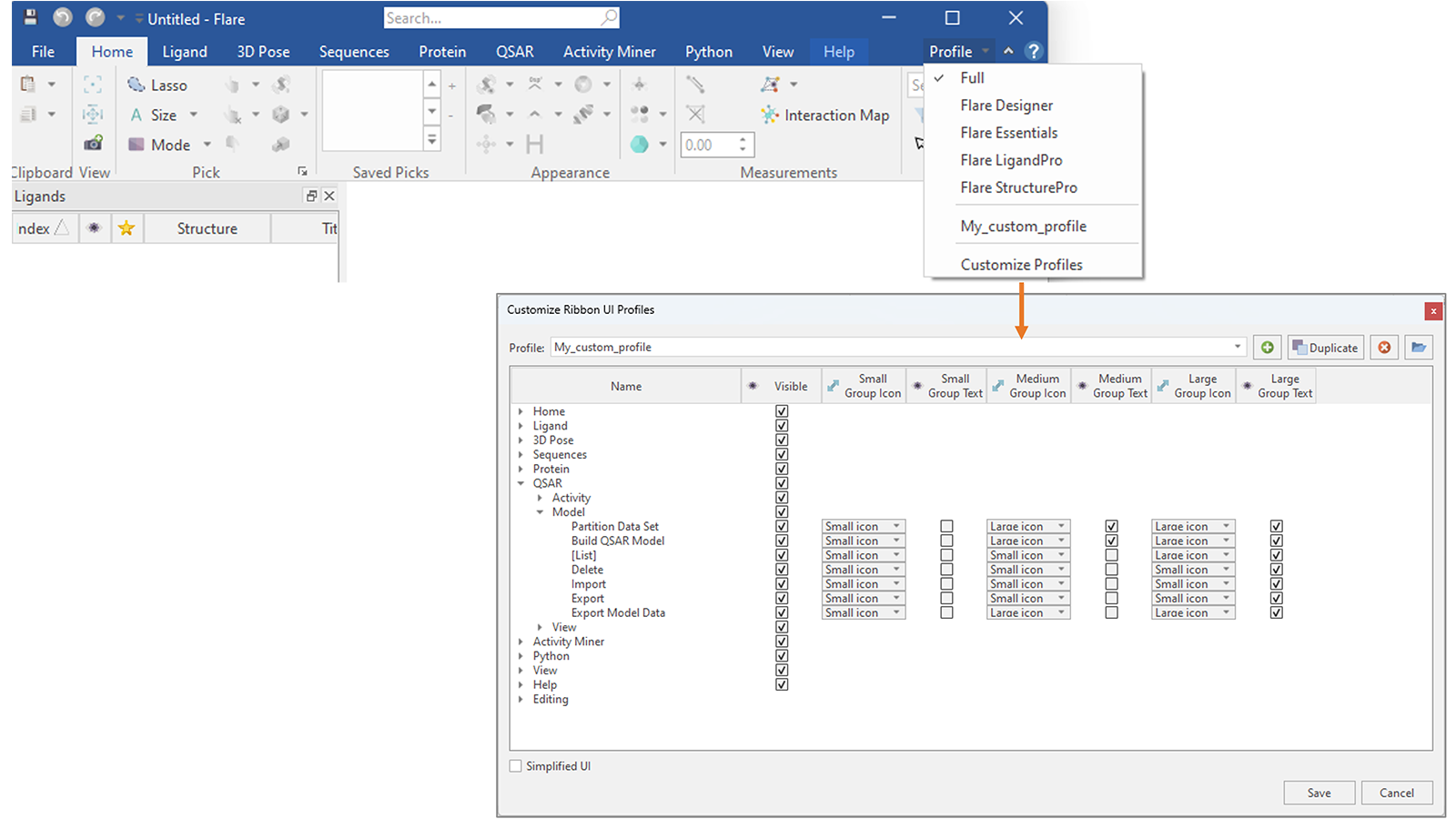
图18. 在Flare V8中,您可以创建自己的自定义配置文件,决定要在Flare菜单中显示哪些选项卡和按钮,以及更改它们的外观。
其他的增强与改进
Flare V8中其他的增强与改进还包括:
- 为共价抑制剂对接补充了更多的弹头
- 对接和打分新增高级选项:指定一个子结构(例如来自共结晶配体构象)以在对接实验期间限制输入构象
- 配体表单中的子结构过滤新增“Is/Is Not”逻辑,能够精确匹配搜索的结构
- 自定义蛋白质飘带的厚度和宽度
- 将肽和蛋白质生长为β折叠或α螺旋
- 选择一个或多个列用作散点图注释标签
- “Column & Activity Editor“新增“Update RDKit Descriptors”功能,用于重新计算导入的RDKit描述符
- 新增“Group By”功能,根据配体表单中的列值对所有配体进行分组
- 在Flare中打开Spark V10.7项目
- Pyflare有新增的脚本与增强
- Flare拓展有新增功能与增强
灵活的软件授权适合于计算化学、药物化学与学术研究
想要在您的项目上试用这些新增强功能和Flare的完整功能,请申请评估并立即试用Flare V8。Flare提供了一系列许可选项,可提供灵活的解决方案,以满足计算化学家、药物化学家和学术研究者的需求。我们的专业支持团队将帮助您入门,在安装和设置期间提供支持,并帮助您访问Flare的各种功能。您将在评估过程中获得全面支持,同时可以自由发布产生的结果并将其用于进一步的研究。
文献
- S. Genheden et al., The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discov. 2015, 10, 5, 449-461
- I. Alibay et al., Evaluating the use of absolute binding free energy in the fragment optimisation process, Commun Chem 2022, 5, 105
- C. E. M. Schindler et al., Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects, J. Chem. Inf. Model. 2020, 60, 11, 5457–5474
- https://wuxibiology.com/application-of-lumo-in-nucleophilic-reactions
- https://science.marshall.edu/pricew/computational/ASSIGN_2/lab2.html
- O. J. Melling et al., Enhanced Grand Canonical Sampling of Occluded Water Sites Using Nonequilibrium Candidate Monte Carlo, J. Chem. Theory Comput. 2023, 19, 3, 1050–1062
- M. L. Samways et. al., grand: A Python Module for Grand Canonical Water Sampling in OpenMM, J. Chem. Inf. Model. 2020, 60, 10, 4436-4441
- Shapovalov, M.V. et al., A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions, Structure 2011, 19, 844-858