LigandScout概述
LigandScout是Inte:Ligand公司的旗舰产品,一款集3D-药效团模型识别、虚拟筛选、精确虚拟筛选的集成平台。它提供了无缝的工作流;支持基于配体或基于结构的药效团建模;还包含了一个全新的高性能叠合算法,其具有前所未有的筛选速度与高品质预测质量。此外,它还提供了用户友好的筛选分析工具,包括自动生成ROC曲线以便进行性能评估。所有的功能都可以通过我们精心设计的图形用户界面来使用,该界面是我们多年的为建立最有人性化的药效团建模工具经验的结晶。本算法以我们在药效团研究领域信誉卓著的知识为基础并经过科学验证,是本领域最先进的技术。
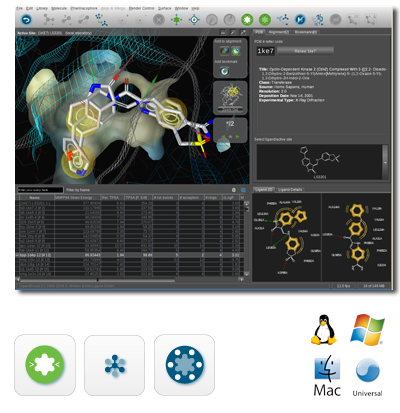
Liganscout用户界面,其支持用见操作系统Linux、Mac与Windows
Ligandscout软件的算法已经在科学杂志上公开发表[1-4]并以我们多年在药效团建模的经验为基础开发了最为先进的药效团应用软件。LigandScout支持多种常用的药效团格式,可以导出模型给Catalyst (Discovery Studio)、MOE与Phase (Schrodinger)等软件进行虚拟筛选以保证最大的协同工作能力。功能齐全的3D图形用户界面支持多层级撤销(undo),使得复杂地活性位点分析与药效团建模变得高效、简单明了。我们设计了结合位点分析、基于药效团的叠合以及共有药效团特征的创建等应用使得LigandScout成为基于结构设计与虚拟筛选的必备工具。Ligandscout还支持所有常见的操作系统。
Ligandscout自成一体,无需任何第三方软件可以完成基于结构与基于配体的药物设计
算法文献
[1] Wolber, G.; Dornhofer, A. A.; Langer, T.; Efficient overlay of small organic molecules using 3D pharmacophores J. Comput. Aided Mol. Des.; 2007; 20(12); 773-788.
[2] Wolber, G.; Langer, T.; LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model; 2005; 45(1); 160-169.
[3] Wolber, G.; Kirchmair H.; Langer, T.; Structure-Based 3D Pharmacophores: An Alternative to Docking? Plenary talk at the 7th International Conference on Chemical Structures in Noordwijkerhout, 2005
点击下载PDF文件:Presentation as PDF file [2.8 MB]
[4] Wolber G., Dornhofer A. A., Langer T.; Efficient Overlay of Molecular 3D Pharmacophores. Oral presentation at the spring ACS meeting in Atlanta, GA, USA, 2006
点击下载PDF文件:Presentation as PDF file [8.8 MB]
主要特性
基于结构的3D药效团设计
- 自动PDB配体识别。
- 最先进的用户界面,支持高级3D图形与撤销功能。
- 2D视图与分级视图直接与3D界面链接。
- 快速将分子生物活性构象叠合至其它分子以及3D药效团,比如从几个配体和或几个药效团生成的3D药效团、公共特征药效团,可以帮助理解、建立合理的作用机制模型。
- 先进的辅酶、离子、水分子以及共价结合配体的处理技术。
- 有经验的用户可以通过丰富的参数进行计算控制。
- 先进的PDB配体识别技术与活性位点配体修复技术。
- 支持将辅酶(co-factor)与水分子设定为配体或生物大分子的一部分。
- 直观地进行基于药效团的分子叠合。
- 支持多种文件格式。
- 对已经编辑过、存储过的结合位点、分子与药效团进行精细准确的管理。
- 智能的互变异构体枚举。
- 支持共价键、卤键、金属配位键等药效团特征(元素)及其虚拟筛选。
精确、快速的虚拟筛选
- 高性能、精确的虚拟筛选,支持自动用ROC曲线进行性能分析、富集因子(enrichment factor)计算。
- 通过工作流,轻而易举地进行靶点与非靶点药效团模型的布林组合,实现多目标同时筛选。
- 用精确的MMFF94力场生成高质量分子结构
基于配体的药效团设计
- 基于配体的药效团建模,包括自动化学特征分类,分配特征权重值,生成排斥体积球,通过基于药效团聚类分析进行全自动选择训练集。
分子对接
- 内置了AutoDock 4.2
- 内置了AutoDock Vina
分子动力学轨迹分析
- 从药效团尺度分析分子动力学轨迹
- 为轨迹的每一帧快照生成药效团模型,您可以比较模型、分析模型
只有蛋白而没有配体-受体复合物结构?
- 可以定义结合位点,生成药效团模型
其它打分函数
- 结合亲和力预测:用Hyde Score精确预测配体与受体的结合亲和力
- 分子形状相似性打分:比较数据库化合物与參比化合物的Gaussian分子形状相似性
- MMFF94结合焓的计算:用MMFF94力场计算化合物与受体之间的结合能
版本与特性比较
| 特性 | LigandScout Essential | LigandScout Advanced | LigandScout Expert/Knime |
|---|---|---|---|
| 基于结构的药效团建模 | |||
| 基于配体的药效团建模 | |||
| 虚拟筛选 | |||
| 模型验证(自动ROC曲线绘制) | |||
| 分子表单的过滤与导出 | |||
| 高性能3D构象搜索 | |||
| 互变异构体生成 | |||
| 基于药效团的聚类 | |||
| 基于药效团的叠合 | |||
| 分子对接 | |||
| 蛋白口袋药效团识别 | |||
| 分子动力学轨迹导入,分析 | |||
| 预先准备好的3D虚拟筛选数据库 | ✔ | ||
| KNIME界面 |
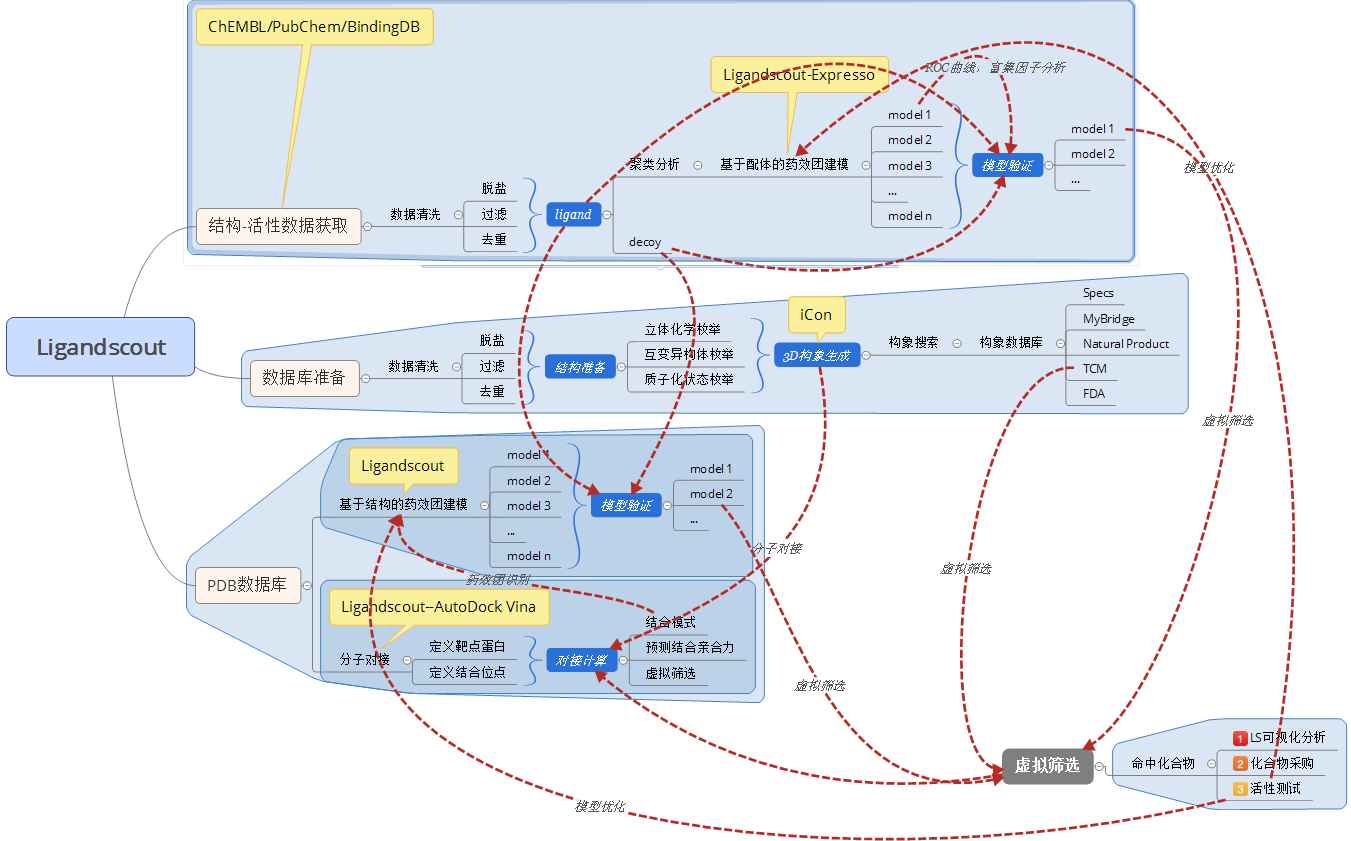
其中,Ligandscout的KNIME具有Ligandscout的全部功能,并且还增加了活性谱预测的节点,见下图。
| Ligandscout Knime节点 | Ligandscout Knime I/O |
|---|---|
 |
 |
Ligandscout Knime界面,不仅包含了Ligandscout的全部功能还新增了活性谱预测节点:将一个化合物与多个药效团模型比较预测可能的生物学活性。
利用Activity Profile节点,与收集到的靶点药效团模型结合,很容易搭建一个靶点预测工作流。下图就是用Ligandscout的KNIME节点与墨灵格靶点数据库结合用来预测化合物靶点的一个例子,在这个例子里,KNIME会自动生成一个靶点预测结果的报告。
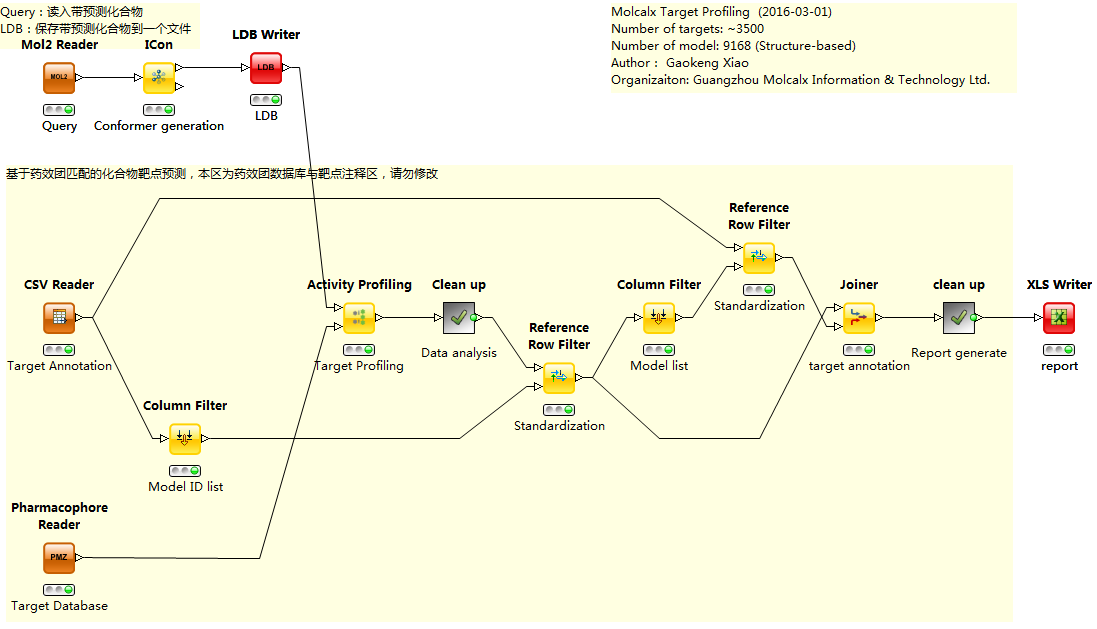
利用Ligandscout的Activity ProfileKNIME节点建立的化合物作用谱分析(靶点预测)工作流
利用Ligandscout与墨灵格靶点数据库进行靶点预测生成的报告样稿
案例分享
1. 基于结构的虚拟筛选发现新的碳酸酐酶VII型抑制剂
文献来源
De Luca, L.; Ferro, S.; Damiano, F. M.; Supuran, C. T.; Vullo, D.; Chimirri, A.; Gitto, R. Structure-Based Screening for the Discovery of New Carbonic Anhydrase VII Inhibitors. Eur J Med Chem 2013, 71C, 105–111.
摘要
在哺乳动物碳酸酐酶(Carbonic Anhydrase,CA)的诸多同工酶中,hCA VII主要表达于大脑并与多个神经生理与疾病相关。已经证实hCA VII是一个有潜力的靶标,选择性的抑制剂可用于治疗癫痫和神经性疼痛。为了发现碳酸酐酶抑制剂(Carbonic anhydrase inhibitors, CAIs)全新的化学实体,作者用了一种基于结构的方法:借助于LigandScout软件从两个著名的CAI于hCA VII复合物晶体结构开始建立药效团模型,获得合并后的药效团模型。随后,用这个药效团模型筛选集中库,将感兴趣的命中化合物进一步对接到hCA VII的晶体结构中。结果识别出几个新的化合物,它们显示出nM水平的CA抑制活性。
用到的软件与关键技术
LigandScout的药效团识别、药效团叠合与合并技术,GOLD的分子对接计算。
2. 基于结构的虚拟筛选发现全新DNA促旋酶抑制剂
文献来源
MatjažBrvar, Andrej Perdih, Miha Renko, Gregor Anderluh, Duš an Turk, Tom Solmajer. Structure-Based Discovery of Substituted 4,5’-Bithiazoles as Novel DNA Gyrase Inhibitors. Journal of Medicinal Chemistry. 2012, 55, 6413-6426.
摘要
细菌DNA促旋酶是一种久经检验并得到确认的新型抗菌药的靶标。它对细菌的DNA复制周期起到关键作用,却不存在于哺乳类动物。这使得细菌DNA促旋酶成为了发展具有选择毒性的抗菌药物的良好靶标。DNA促旋酶B亚型的ATP结合位点非常重要,并已有广泛研究。为了发现细菌DNA促旋酶B亚型ATP结合位点的全新抑制剂,作者采用了两步设计策略:第一阶段中使用LigandScout软件分析CBN(一种香豆素类抑制剂)与G24蛋白(包含ATP结合位点的N端最小的片段)之间的相互作用,构建药效团模型进行虚拟筛选,然后用分子对接方法进一步筛选;第二阶段,对第一阶段识别出来的4,5’-二噻唑结构类型构建集中库,采用对接方法进行筛选。抗菌活性测试结果确证了几个化合物具有DNA促旋酶B型的抑制活性,对其中最好的抑制剂还采用SPR、MST、DSF及X-ray等多种实验技术进行结构表征。
亮点
- 采用两步设计策略:先以天然抑制剂的结构数据为基础进行筛选,再以第一阶段的苗头化合物建立集中库进一步筛选;
- 对药效团命中的化合物进一步进行分子对接计算;
- 采用LigandScout基于结构的药效团建模技术与GOLD分子对接方法,可发现全新骨架的抑制剂;
- 先后发现9个化合物具有低微摩尔水平的抑制活性。
- 采用多种生物物理和结构生物学技术,揭示了抑制剂的结构特征与结合模式。
3. 基于配体的药效团建模与虚拟筛选发现全新17β-羟基类固醇脱氢酶2抑制剂
文献来源
Anna Vuorinen et al. Ligand-Based Pharmacophore Modeling and Virtual Screening for the Discovery of Novel 17β -Hydroxysteroid Dehydrogenase 2 Inhibitors. J. Med. Chem. 2014, 57, 5995 −6007.
摘要
17β-羟基类固醇脱氢酶2(17β-HSD2)催化雌二醇到雌素酮的失活转化,对它的抑制可作为治疗骨质疏松症,改善雌激素缺乏症的治疗选择。作者使用LigandScout构建了17β-HSD2抑制剂的药效团模型,进行虚拟筛选,生物实验证明29个苗头化合物中有7个具有低微摩尔级IC50活性。为了进一步研究其构效关系(SAR),从SPECS数据库中又挑选了30多个衍生物用于测试。最终发现3个活性最好的化合物,最低活性达240nM。还发现了1个对17β-HSD1具有选择性的抑制剂,3个对其他相关HSD具有选择性,6个对其他HSD没有影响。
亮点
- 采用多种药效团组合虚拟筛选策略:以4个不同结构的抑制剂为基础,构建3个药效团模型,进行组合筛选。避免因结构差异过大而产生公共特征过少或合并特征过多的药效团模型,导致筛选结果失去意义;
- 通过测试集检验结果对自动化产生的模型进行调整,包括:去除部分特征、调整排除体积大小以及设置可选特征;
- 采用多条件过滤,对药效团筛选结果进行浓缩,包括:改良的Lipinski规则、聚类分析、化合物性质预测(类药性、诱变性、刺激性以及致癌性)。
- 构效关系研究:对第一轮筛选结果进行配体扩张,结合简单的2D结构相似度与药效团模型,搜索全新骨架的衍生物,进行构效关系研究;
- 根据构效关系研究的结论,进一步调整药效团模型,提高特异性与敏感性。
- 进行多靶标活性测试,获得选择性良好的化合物。
4. 药效团建模、虚拟筛选及体外测试揭示氟哌啶醇、依普拉酮及芬布酯是神经激肽受体配体
文献来源
Yvonne Krautscheid et al. Pharmacophore Modeling, Virtual Screening, and in Vitro Testing Reveal Haloperidol, Eprazinone, and Fenbutrazate as Neurokinin Receptors Ligands. J. Chem. Inf. Model. 2014, 54, 1747 −1757.
摘要
神经激肽受体(Neurokinin receptors,NKRs)被报道参与多种生理过程,因此是有前景的药物靶标,但是它同时也可能是许多药物的副作用的诱因。作者在亚型选择性和非选择性NKR拮抗剂的基础上,使用LigandScout构建了一组基于配体的公共特征药效团模型。使用该模型对已批准药物进行虚拟筛选>。该前瞻性研究结果得到体外实验的证实,他们发现氟哌啶醇、依普拉酮及芬布酯是NKR的配体。该研究表明,药效团模型不仅用于常规的药物发现,还可用于建立化合物的活性谱,同时本文也是药效团在老药新用开发的典范。
亮点
- 通过精心准备的数据库的检验,从众多药效团模型中挑选命中结果较少重叠的一批药效团,进行组合模型筛选,以达到更大的命中覆盖面;
- 通过药效团模型筛选的方法,从老药中寻找新功能(副作用)。
5. 基于药效团片段的虚拟筛选
文献来源
Deyon-Jung, L., et al. (2016). “Fragment pharmacophore-based in silico screening: a powerful approach for efficient lead discovery.” MedChemComm 7(3): 506-511. DOI:10.1039/C5MD00444F
摘要
摘要:欧莱雅公司的研究人员对已经批准上市的美容用分子进行碎片化、官能团化与重新组合等一系列处理,构建了适于虚拟筛选的虚拟库。对该虚拟库进行了基于药效团的虚拟筛选以及三个月的优化阶段后,发现了一个各种性质都理想的先导物,其有望在美容应用领域里作为皮肤护理的候选分子。本项目的成功为其它美容靶点的先导物发现铺平了道路。
亮点
- 高质量的虚拟组合库准备方法:将上市批准的化妆品分子碎片化为精选的片断,再将片断按照特定规则进行重组生成初始的92000化合物库,这些化合物适合于化妆品使用的目的;
- 片段化药效团虚拟筛选:首次实践将一个大的药效团拆解为两个小的药效团进行虚拟筛选;
- 苗头化合物的扩增全程获得模型支持,快速发现了10μM水平的先导物。
感兴趣的靶点是一个皮肤蛋白,首先用Ligandscout基于配体-蛋白相互作用生成药效团模型,这个模型包含了全部的配体-受体相互作用。然而这个模型的两个最远药效团元素之间距离特别大(达到15Å),而数据库的分子并没有那么大的尺寸,因此考虑采用片断化的药效团进行虚拟筛选:将初始的药效团人为的分为三个部分,组合成成两个小药效团模型去虚拟筛选数据库。
6. 基于结构的虚拟筛选发现苹果蠹蛾信息素
文献来源
Liu, J., et al. (2016). “Structure-based discovery of potentially active semiochemicals for Cydia pomonella (L.).” Scientific Reports 6: 34600. DOI:10.1038/srep34600
摘要
摘要:信息素对昆虫行为的控制具有非常重要的意义,但信息素的开发是一个费事、费力的研究过程,因此如何高效地筛选信息素具有非常重要的意义。本文分享了刘吉元博士用Ligandscout进行虚拟筛选发现信息素的案例:通过药效团建模、虚拟筛选、基于分子形状的打分、结合亲合力打分等多步过滤后从数据库中富集潜在的苹果蠹蛾性信息素结合蛋白1(Cydia pomonella pheromone binding protein 1, CpomPBP1)配体,最后进行生物活性测试发现4个活性化合物。
亮点
- 先虚拟筛选富集活性化合物,再生物活性测试:尽量少的实验发现活性化合物,大大提高了信息素的发现效率;
- 使用多种打分策略,虚拟筛选的每个步骤意义清晰、明确,为虚拟筛选在信息素活性化合物发现提供了简单易用的成功案例、值得推广
系统要求
内存:至少2GB的内存,推荐4GB或以上
操作系统:Windows 7,Windows 10,MacOS X,需要安装JRE (Java runtime enviroment).
显卡:支持OpenGL 1.2的硬件加速3D显卡(推荐NVIDIA或ATI)
CPU:即使是便宜的单机版用户,也支持单机多核的并行,无需添加License
访问Inte:ligand公司网站
Inte:ligand网站:http://www.inteligand.com