![]()
Lead Finder™
Great leads from high-throughput docking
概述
Lead Finder设计的目的是用来满足计算化学家和药物化学家在药物发现过程中的需求,以及药理学家和毒理学家在计算建模、评估ADMET的需求、以及从事酶专一性和合理酶设计的生物化学家和酶学家的需要。
Lead Finder可以用于:
- 通过虚拟筛选发现全新的先导化合物
- 通过虚拟筛选对活性化合物的高富集能力设计集中库
- 预测活性化合物的3D结构
- 共价或非共价对接计算
- 快速地评估新设计的分子是否与结合位点匹配
操作系统与使用方式
Lead Finder通过Linux,Windows与Mac OS命令行界面进行使用。
Lead Finder的软件组成
Lead Finder软件包包含build_model与leadfinder两个子程序:前者负责蛋白结构准备;后者负责计算能量格点与分子对接。
Build Model是Lead Finder里准备蛋白的计算模块,它根据Mehler E. L.的SCP理论1,2在指定的pH值下归属蛋白残基离子化状态、准备蛋白结构,包括:(1)根据给定的pH值优化残基的离子化状态对蛋白加氢;(2)根据出现在蛋白里的配体、辅酶、底物优化极性氢的取向;(3)优化His、Asn与GLn残基的侧链取向;(4)残基的侧链重建;(5)氨基酸残基突变;(6)单个残基GAP的填充。
Lead Finder操作步骤
Lead Finder分子对接分3个步骤:(1)蛋白结构准备;(2)生成格点文件;(3)分子对接。
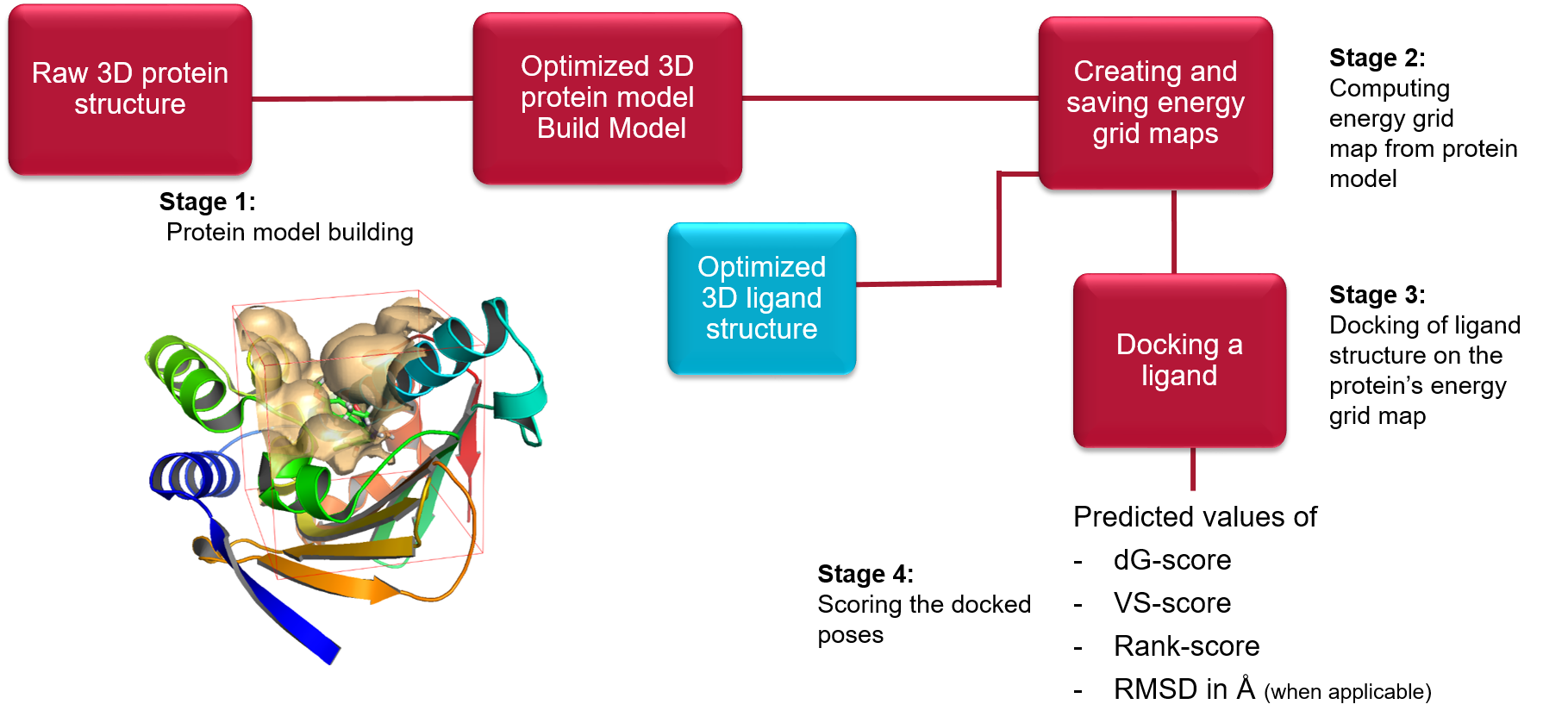
Lead Finder分子对接操作流程
Lead Finder分子对接的成功率
分子对接的精度用对接的成功率来评估。将从蛋白-配体复合物提取的配体坐标作为参比,如果得分最高的pose与参比配体相比的RMSD<2 Å,则认为重现结合模式,对接正确。对接成功率是指针对一组配体-蛋白复合物结构成功对接的百分比。虽然对接成功率的还有其它的定义,但这里使用了最广泛接受的定义以便使Lead Finder基准与别处获得的竞争性软件基准具有可比性。另外,为了增加重现性,所有Lead Finder对接计算独立地执行20次,并且只有当以2 Å或更好的精度生成最高排名pose的概率至少为50%时,对接才被认为是成功的。
跟据测试,Lead Finder比同行GOLD,GLIDE,FlexX, Surflex-Dock在同样的数据集上显著地表现出更加优秀的结合模式预测性能(re-docking精度)。
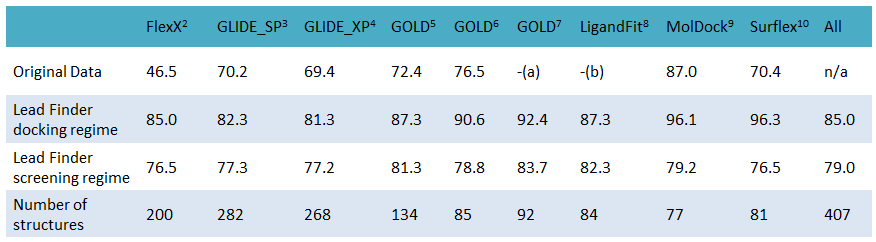
上表数据出处:BioMolTech,Original Data行为对应列软件的对接精度;Lead Finder行的每列对应着Lead Finder用相应软件测试集测试获得的性能结果。
Lead Finder分子对接的虚拟筛选性能
Lead Finder的虚拟筛选性能用34哥蛋白靶标评估过,这些靶标的选择标准是:
- 该蛋白适合于药物发现研究
- 具有高质量的3D结构
- 具有足够数量的、已知的配体
对于每个蛋白靶标,从以下公共来源提取一组活性配体:PDB数据库、KiBank、Surflex开发者公开的活性配体。在所有当前虚拟筛选研究中使用的1904个诱饵配体是从最初的Surflex出版物中借用的,可从Surflex开发者网站下载。
基准实验包括创建化合物的测试化合物库(将活性配体与一组诱饵(Decoy)化合物混合而得)、将数据库的每个化合物与特定靶标对接、以及根据计算的VS打分值(score)对化合物进行排序。为了定量表征虚拟筛选效率,使用了两个公认的参数:ROC曲线下面积(AUC)和富集因子(EF)。虚拟筛选实验的结果列于下表中。
数据来源: BioMolTech
Lead Finder的方法学文献
- Stroganov O.V. et al., Lead Finder: an approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening, J Chem Inf Model. 2008 Dec;48(12):2371-85. PubMed ID: 19007114
- Novikov F. et al., CSAR scoring challenge reveals the need for new concepts in estimating protein-ligand binding affinity, J Chem Inf Model. 2011 Sep 26;51(9):2090-2096. PubMed ID: 21612285
- Novikov F.N. et al., Improving performance of docking-based virtual screening by structural filtration, J Mol Model. 2010 Jul;16(7):1223-30. Epub 2009 Dec 30
- Novikov F.N. et al., Lead Finder docking and virtual screening evaluation with Astex and DUD test sets, J Comput Aided Mol Des. 2012 Jun;26(6):725-35
- Zeifman A.A. et al., Hit clustering can improve virtual fragment screening: CDK2 and PARP1 case studies, J Mol Model. 2012 Jun;18(6):2553-66. Epub 2011 Nov 9
- Stroganov O.V. et al., TSAR, a new graph-theoretical approach to computational modeling of protein side-chain flexibility: Modeling of ionization properties of proteins. Proteins, 2011 Sep; 79(9):2693-2710. PubMed ID: 21769942