水分子的处理非常重要
所有的生物学过程都发生在水环境中,当小分子-大分子相互作用时,水分子起到非常重要的作用,因此必须要正确处理这些水分子。比如,分子对接准备受体结构时,什么样的水要保留做为受体的一部分? 什么样的水要删除?正确的决策非常重要,直接影响了后续工作。
SZMAP是OpenEye应用软件包中的一个模块,是一款水分子位置与稳定性预测软件软件[1]。SZMAP采用显式水分子探针在高介电连续溶剂(high-dielectric continuum solvent)中快速计算分子表面附近溶剂化自由能的大小与分布。我们把这种方式称为半连续模型(semi-continuum model)[2, 3],它可以克服连续模型的缺陷,比如波松-玻尔兹曼方法[4]。采用半连续泊松-波尔兹曼理论来定量绘制与配体结合状态态的受体(Holo受体)、非配体结合状态的受体(Apo受体)和配体结构的热力学量变图,它可以预测特定水分子对配体结合的稳定化或不稳定化效应,还可以识别结合水的位置和哪些位置的水是无序的。在结合位点,更好地理解这些效应有助于先导化合物的优化和药物设计其他方面问题的研究。比如,在对接研究中,可以分析哪些水分子在对接时应该保留并当成受体的一部分,哪些水是应该被删除掉的。
SZMAP:分析配体-受体相互作用时水分子的作用
SZMAP (solvent-Zap-mapping)[1]是一种半显式溶剂映射方法,可用来研究蛋白-配体复合物中水的热力学性质,它兼具了显式与隐式溶剂模型方法的优势。如图1所示,SZMAP将一个显式水探针放入到蛋白-配体体系里,水探针是一个三原子组成、与许多全原子力场的水模型相似,具有偏电荷(partial charges)。而其余的溶剂用ZAP toolkit的Poisson-Boltzmann隐式溶剂模型表示。SZMAP可对放置于蛋白上网格的每个点采集水的能量,从而在结合位点里构建水的热力学结合参数的三维映射图;当然也可以仅对用户指定的坐标点进行计算。比起显式的溶剂方法,SZMAP显而易见的一个优势是计算速度,因为它避免了为了获得可靠结果而进行长时间的平衡与高强度采样。

图1. SZMAP在蛋白-配体研究体系里放置显式水分子探针,并用隐式水填充其它位置。SZMAP可以在格点上或用户指定的位置采集水的取向
SZMAP既可在每个格点上、也可以在指定的位置上放置探针计算热力学性质。在给定的探针位置,SZMAP采集探针水的各种取向。对于每一个取向,计算体系的能量(Ej),包含下面几个成分:水探针与蛋白以及配体的vdW相互作用(Evdw protein-water),水探针与配体、蛋白的静电相互作用以及极化的PB连续介质(溶剂)的相互作用(EPB protein-water),还有蛋白、水的静电去溶剂化项(Epsolv与Ewsolv)。
(1) 
水探针取向之系综及其能量可以用来构建配分函数Q:
(2) 
从配分函数Q,SZMAP可以推导出探针处于采样的某一个方向时的概率Probj:
(3) 
概率与配分函数可用于SZMAP的热力学性质计算,比如与连续介质的水比较计算探针水取向的熵变(Eq 4),探针的自由能变化(∆G, Eq 5)以及焓变(Eq 6):
(4) 
(5) 
(6) 
此外,SZMAP还可用来计算水探针相对于两个不同参比态的热力学性质(图2)。其中一个参比态为真空探针,是一个置换了连续介质溶剂的球形空腔,与水探针相似却不含任何原子,对vdW能量项没有任何贡献。这个参比态设计用来在同一个位置对水探针与真空空间进行比较。另一个参比态是中性水探针(neutral water probe),它是对脂肪烃基的模拟。中性水探针与水探针一样由三原子组成,但是所有原子的偏电荷均为0。此外,对中性水探针进行与对水探针一样的取向采样。因此,当中性水探针的自由能减去水探针的自由能,就抵消了vdW项,那么能量差就反映了由于水分子偏电荷带来的结合能差异。
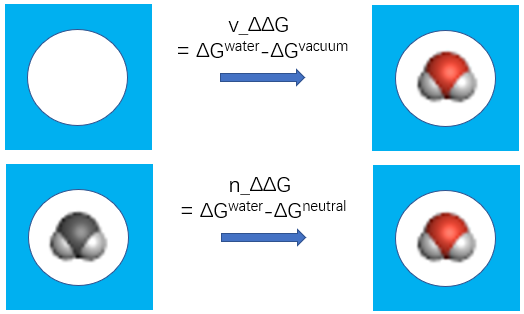
图2. SZMAP的参比态示意图。上:真空参比态;下:中性参比态。水探针的着色反映了氢氧原子的偏电荷,而中性参比态没有着色反映了这些原子是没有电荷的。
SZMAP的一个主要应用,如其名所暗示,是在蛋白结合位点里映射出水的偏好位置。SZMAP可以在蛋白-配体复合物的网格点上进行采样,计算的溶剂化自由能以及其它热力学性质项的等值面等经渲染后可用来可视化呈现结合位点内这些值的变化,SZMAP的水-中性探针自由能差(neutral probe free energy difference,简称n_ddG或n_ΔΔG)衡量了正常、带电的水探针比不带电荷而具有水形状探针的偏好程度。在结合位点里,如果n_ddG数值为负数则表示在亲水区有利,如果数值为正则表示在疏水区有利。如果为0或接近0,意味着对极性或非极性分子均无偏好。一般来说-0.5kcal/mol的等值图可以用来将水分子探针比非极性水探针显著有利的位置给识别出来。
稳定化自由能:水对蛋白-配体相互作用的影响
当蛋白-配体复合物结构可获得的时候,我们可以用SZMAP进行一个特殊格点计算,即稳定化自由能(stabilization free energy):
(7) 
其中bulk-溶剂的自由能定义为0.0kcal/mol。稳定化自由能评估了蛋白-配体结合对水能量的影响。
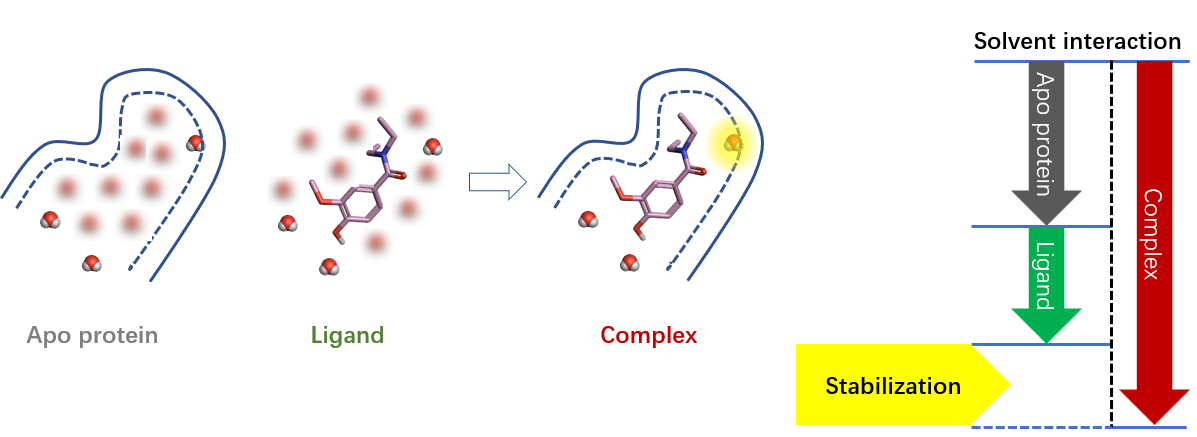
图3. 负的稳定化自由能(稳定化)形成示意图
如图3所示,负的稳定化自由能区为结合对水有稳定作用的区域,或者换个角度看,水的存在可以稳定蛋白-配体的结合(有助于蛋白-配体的结合)。对于这样的水分子,最简单的处理方式是留着不要动它,因为它们可以提高结合亲和力;还可以用一个起到相似相互作用的基团对水分子进行替换,以便保留相互作用而不损害结合亲和力。
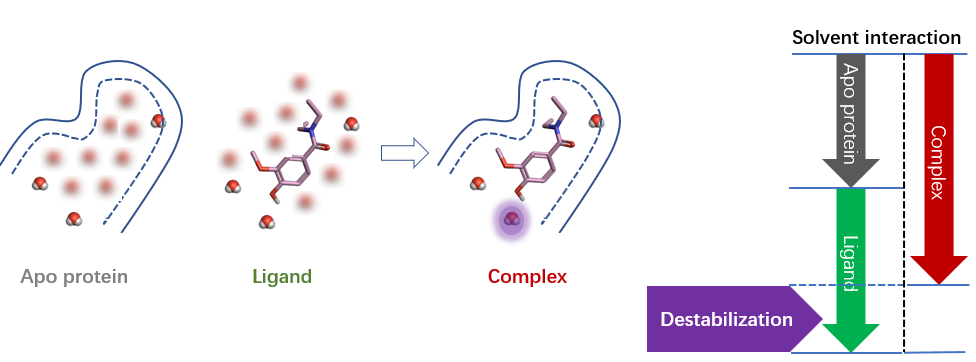
图4. 正的稳定化自由能(去稳定化)计算示意图
如图4所示,正稳定化自由能表示在该区域水对蛋白-配体的结合起到去稳定化的作用(对蛋白-配体的结合不利)。正稳定化自由能的水使蛋白-配体结合亲和力降低,去稳定化区域的水也相对容易有效地进行置换。还可能通过配体修饰,改善与去稳定化水的相互作用(甚至因此逆转为稳定化的水)或者替换去稳定化水提高结合亲和力。
总的来说,SZMAP的Stabilization计算对如何处理配体周围水的提供了SAR指导:在stabilization区可以保留水或模拟水的作用;而在destabilization区可以通过置换(displace water)或改善静电作用来实现化合物的优化。
中性探针自由能差(neutral probe free energy difference)
(8) 
方程8给出了SZMAP水-中性探针自由能差(neutral probe free energy difference,简写为n_ddG或n_ΔΔG)的定义,它衡量了正常、带电的水探针比不带电荷而具有水形状探针的偏好程度。如图5所示,在结合位点里,如果n_ddG数值为负数则表示在亲水区有利,如果数值为正则表示在疏水区有利。如果为0或接近0,意味着对极性或非极性分子均无偏好。一般来说-0.5kcal/mol的等值图可以用来将水分子探针比非极性水探针显著有利的位置给识别出来。
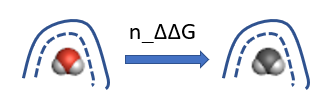
图5. 中性自由能差(n_ddG,n_ΔΔG)计算方法示意图。左:彩色的水是正常水,右:黑白的水是不带电荷(中性)的水。
请注意,不要混淆稳定化自由能与n_ddG,两者在数值或符号上不总是相关的(可以不相关):在复合物中的中性差自由能(n_ddG,n_ΔΔG)为负的区域中可以找到正稳定化自由能,反之亦然。两者通常结合使用:稳定化自由能给出水的处理方法,而n_ddG给出SAR并用于指导分子设计。
用稳定化自由能解释HSP90抑制剂的SAR
Kung等人[5]靶向结晶水的Hsp90抑制剂发现策略为水分子计算提供了非常好的素材,在该算例中涉及图6所示的6个化合物1-6。
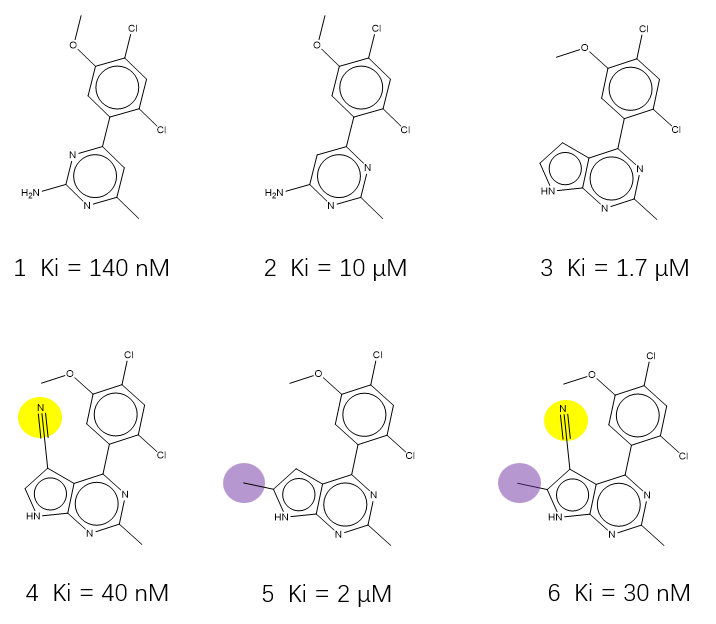
图6. Kung等人靶向结晶水的Hsp90抑制剂算例化合物。黄色高亮:W1水replace基团;紫色高亮:W3水displace基团。
对化合物1与Hsp90的共晶复合物关结构的结合位点进行稳定化格点计算,结果如见图7所示,涉及到三个关键结晶水,分别为W1、W2与W3。其中W1与W2的稳定化自由能小于0(分别为-0.71与-1.27kcal/mol),对化合物1-Hsp90复合物结构起到稳定化的作用,也就是对化合物1的结合亲和力有利;而W3的稳定化自由能大于0(为0.024kcal/mol),对化合物1与Hsp90的结合不利,起到去稳定化的作用,该去稳定化自由能ΔΔG值很小接近于0,这也意味着W3对蛋白-配体复合物的影响很小。同时W3附近有个紫色的去稳定化区,在后续设计中应该注意是否需要往该区域延伸。
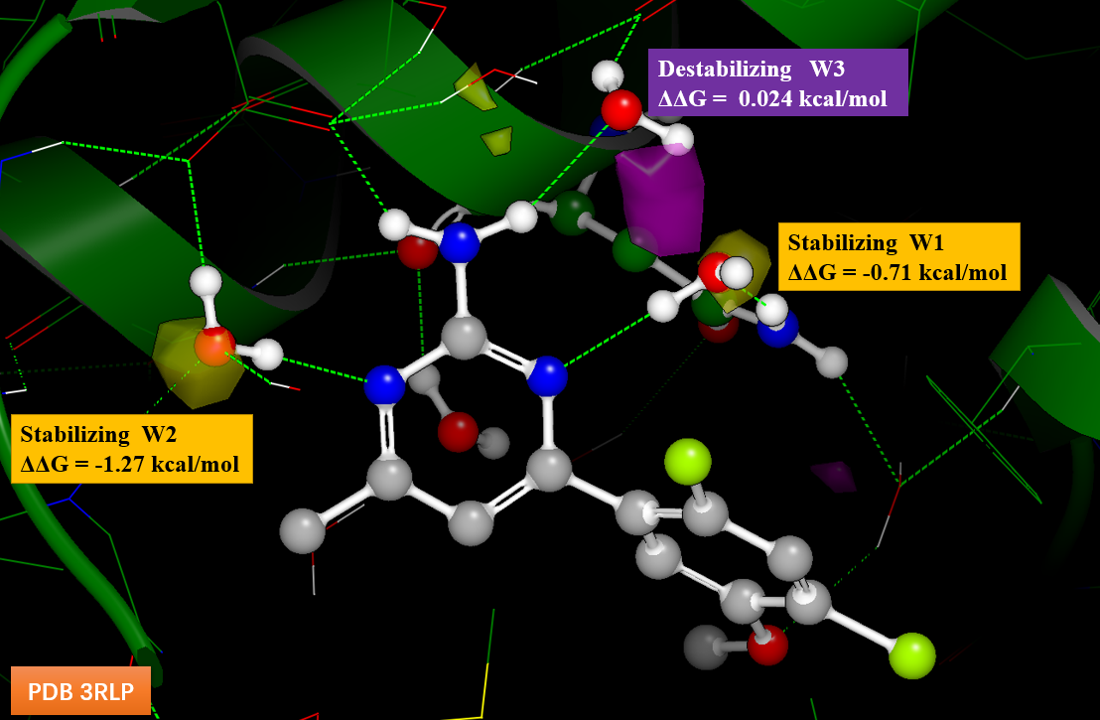
图7. 化合物1-Hsp90复合物结构(PDB 3RLP)的稳定化网格计算
化合物2与1的区别是氮原子的位置,比起化合物1,2不能与W1发生氢键相互作为,因此没有获得W1对2的稳定化作用,活性降低了大约71倍。同样的道理,与1相比,化合物3也没获得W1的稳定化作用,因此活性降低了12倍。这证明了W1水的重要性,这与W1负的稳定化自由能相一致:与W1相互作用有利于结合亲和力,反之相反。
化合物4是在化合物3上引入腈基(图6 4的黄色高亮部分)对W1水j进行替换的设计。化合物4的腈基模拟了W1的相互作用,化合物4比3的活性提高了43倍。这进一步证明了W1水的重要性,与W1负的稳定化自由能相一致,模拟此类水的相互作用进行的替换设计可以提高化合物的结合亲和力。
化合物5是在化合物3上引入甲基(图6 5的紫色高亮部分)对W3进行置换的设计。虽然W3的稳定化作用能大于0,但接近于0,因此可以估计该置换对活性的影响并不大,这与化合物5、3的Ki值相差不大(分别为2与1.7μM)是一致的。
化合物6是在化合物3上同时引入对W3置换的甲基(图6 6的紫色高亮部分)与对W1替换的腈基(图6 6黄色高亮部分),化合物6兼具了4、5设计的优势,其活性比3提高了57倍。6的活性比4略优,但是相差不大,与甲基置换的W3稳定化能接近于0是有关系的。化合物6的设计再次证实起始的假设:对负稳定化自由能的水进行模拟(替换)与对正稳定化水置换的策略是正确的。
应该要注意的是,根据方程7我们可以推知:同一个蛋白结合位点里的同一个水,与不同配体结合的时候,这个水的稳定化自由能是可以不同的!因为方程7的稳定化自由能取决于复合物、apo蛋白与配体。从化合物1-HSP90复合物结构(PDB 3RLP)角度看,W1对化合物1与HSP90的结合是有利的;但是从化合物2的角度看W1,W1就不能稳定化化合物2-HSP90的结合亲和力,此时稳定化自由能就为正!从方程7看,此时我们就需要改造化合物2以提高2与W1的相互作用,从而将正的稳定化自由能变成负的(从2出发设计出1就是这样的过程),这正是稳定化自由能计算给我们的SAR提示。
用n_ΔΔG解释BTK抑制剂的SAR
图8所示的化合物7与8是一对BTK抑制剂[6],其中7对BTK的抑制活性是其甲基化衍生物8的112倍,IC50分别为1.3与145nM。
。
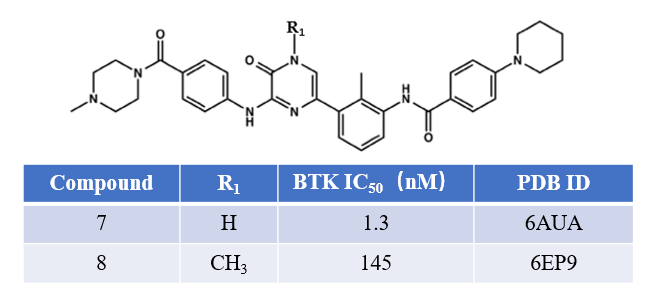
图8. BTK抑制剂化合物7与8的化学结构及其活性[6]
结构生物学数据表明,除了与HOH843的相互作用之外,抑制剂7、8在与BTK的相互作用模式几乎一模一样(如图9所示):7与结合位点的HOH843发生氢键相互作用(PDB 6AUA),而化合物8的甲基则置换了该水分子(PDB 6EP9)。
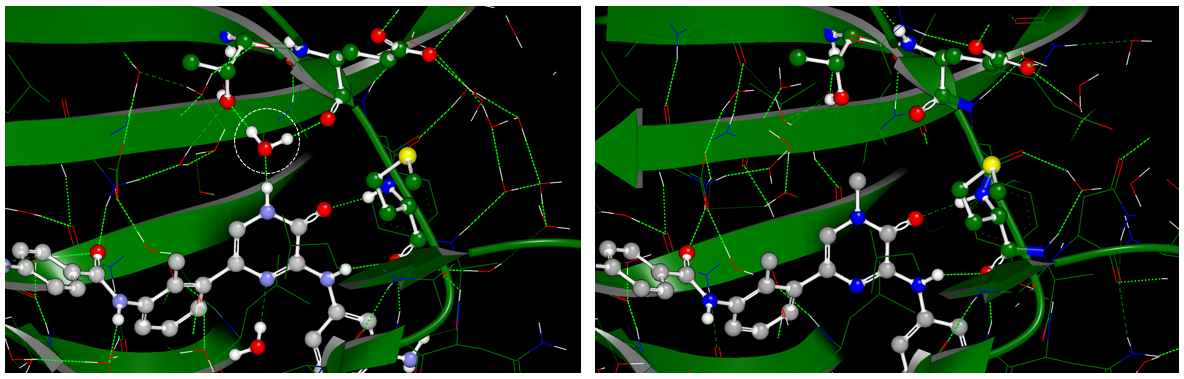
图9. 化合物7、8与BTK相互作用比较示意图。左:化合物7与BTK的结合模式(PDB 6AUA);右:化合物8与BTK的结合模式(PDB 6EP9);左边白色圆圈的水为HOH843,在6AUA中与化合物1发生氢键相互作用,在6EP9中被化合物2的甲基置换而消失。
将PDB 6AUA与6EP9按Cα进行重叠,发现化合物7与8基本完全重合,所以这一对分子在构象能上也没有差异,其生物学活性活性差异可以归结为对水分子HOH843的置换带来的,这为水分子置换计算解释SAR提供了非常好的算例。
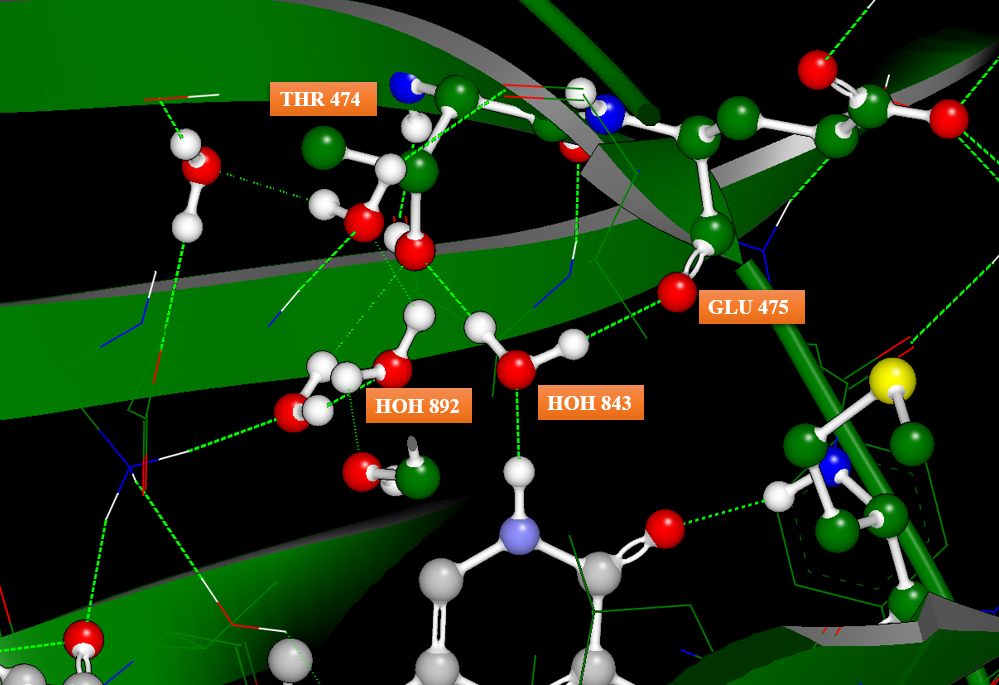
图10. PDB 6AUA的水分子HOH 843通过氢键桥连着配体与蛋白相互作用、介导着氢键网络
图10展示了PDB 6AUA的HOH 843在化合物7结合位点里氢键网络,它通过氢键桥连着配体与残基THR474以及GLU475,并与HOH892、871形成水的氢键网络。形成鲜明对比的是,在化合物8-BTK的共晶复合物(PDB 6EP9)里该水消失,其它的氢键网络水却还保留着,化合物8的活性却下降为1的100多倍。因此,理论预见HOH 843的重要性具有有非常重要的药化指导作用。
鉴于我们感兴趣问题是在化合物7上引入一个甲基以置换结晶水HOH843的分子设计(化合物8)是否对活性有利,因此,用SZMAP对HOH843位置进行n_ΔΔG分析正是我们需要的:如果HOH843处的n_ΔΔG为负值,说明引入甲基对活性不利;如果HOH843处的n_ΔΔG为正值,说明引入甲基对活性有利。
首先从PDB上下载PDB 6AUA,然后用Spruce进行蛋白结构准备,取Chain A的Alternative A用于SZMAP计算stabilization格点:
szmap -mpi_np 24 -stabilization \
-prefix 6auaA_stbl \
-p 6AUA_AaltA__DU__BXJ_A-704.oedu \
-excludeDUsolvent
其中,-stabilization指示szmap进行稳定化格点计算;其中-excludeDUsolvent指示szmap将体系里的水忽略。这一步需要15分钟左右,结果获得一个新文件6auaA_stbl.oeb.gz。该文件包括了我们需要的SZMAP稳定化格点以及各种热力学性质。
还可以将Apo结构里HOH843所在位置的热力学性质提取出来,比如n_ddG:
szmap_grid -input_mol 6auaA_stbl.oeb.gz \
-grid neut_diff_apo_free_energy_grid \
-at_coords HOH843.pdb
结果如下:
note: Attaching values from 'neut_diff_apo_free_energy_grid' to atoms in szmap_grid_interp.oeb.gz tagged as 'szmap_neut_diff_free_energy'.
OpenEye SZMAP results from 'neut_diff_apo_free_energy_grid':
num atom x y z mask n_ddG
1.1 O 19.583 5.022 4.047 1 -2.370
1.2 H2 19.669 5.871 4.484 0 -0.276
1.3 H1 19.240 4.409 4.758 0 0.000
1.total 1 mask > 0.800 1 -2.370
1.avg 1 mask > 0.800 . -2.370
我们可以发现,HOH843氧所在位置的n_ΔΔG = -2.370kcal/mol,因此引入甲基的设计是对活性不利的,这与实验结果一致,活性降低了大约110倍。
用VIDA进行可视化分析,重点是对蛋白Apo结构的结合位点进行“水-中性探针自由能差(n_ΔΔG)分析。这里,我绘制了两个等值图:若n_ΔΔG小于-1.5kcal/mol,则将等值面渲染为黄色,以示水结合有利区(或者是中性水代表的亲脂基团结合有不利区);若n_ΔΔG大于0.8 kcal/mol,则将等值图渲染为紫红色,以示水结合不利区(或者是中性水代表的亲脂基团结合有利区)。
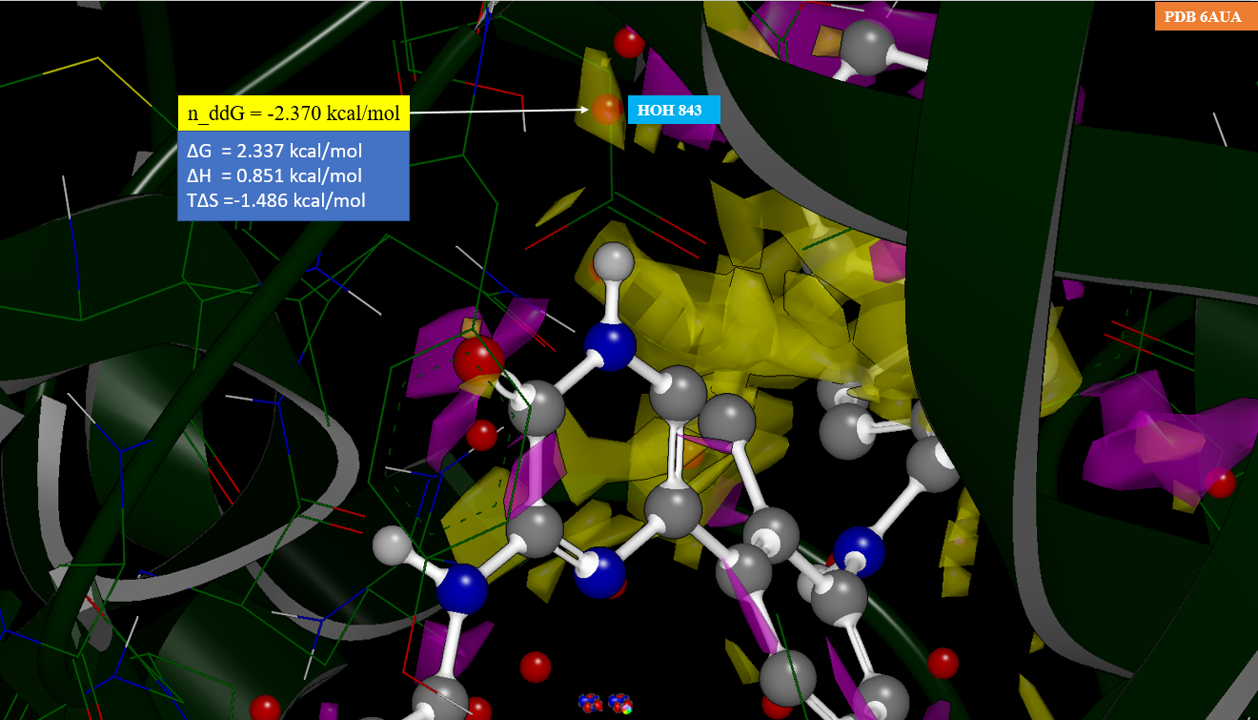
图11. SZMAP计算结果,红色球:结合位点里的结晶水;等值图用中性探针自由能差n_ΔΔG渲染:黄色为n_ΔΔG小于-1.5 kcal/mol等值面,紫红色为n_ΔΔG大于0.8 kcal/mol等值面。
对6AUA结合位点Apo结构进行的n_ΔΔG可视化分析结果如图10所示:黄色区域为n_ΔΔG小于-1.5kcal/mol,紫红色区域为n_ΔΔG大于0.8。与配体酰胺NH相互作用的HOH843位于黄色等值面里,为n_ΔΔG小于0的区域,HOH843的n_ΔΔG = -2.370kcal/mol。
中性探针自由能差(n_ΔΔG)提供了蛋白角度的SAR分析:黄色区域为引入亲水基团有利的区域,而紫色区域为引入亲脂基团有利区,因此可以从配体与蛋白的匹配性来看分子设计是否合理。化合物7的酰胺羰基氧落在n_ΔΔG小于0的黄色区域,说明化合物这部分结构特征对结合是有利的。我们会发现化合物7的很多亲脂片段与紫色网格重合,紫色区域意味着代表脂肪烃基的无电荷水有利区,这意味配体的亲脂性部分与蛋白结构特征匹配良好。同样的道理,用甲基取代酰胺氮上的氢,则亲脂性的甲基与黄色网格代表的极性基团有利区相冲突,因此化合物8的甲基与蛋白结构特征是不匹配、对活性不利的设计。
综合利用稳定化自由能与中性水探针自由能差解释KRAS G12C共价抑制剂的先导化合物SAR
MRTX849 (Adagrasib)是Mariti开发的一种口服有效的、高度选择性的KRASG12C抑制剂。MRTX849通过将KRAS分子不可逆地锁定在其非活性状态来最大化抑制作用,从而防止肿瘤细胞生长并导致肿瘤细胞死亡。与现有的KRAS突变体选择性抑制剂相比,MRTX849的半衰期和进入肿瘤的渗透率显著提高,在整个给药期间中都关停了KRAS信号传导,并显示出更高的抗肿瘤活性。目前,MTRX849处于三期临床研究阶段。

图12. MTRX849优化过程中的两个关键优化步骤
在MTRX849的研究过程中[7],涉及到如图8所示的两步关键优化:1)在化合物7哌嗪环引入腈基,提升了440倍的活性;2)在萘环8位引入了氯,提升了10倍的活性。两次优化总共提升4400倍的活性,但是一共才引入4个原子。
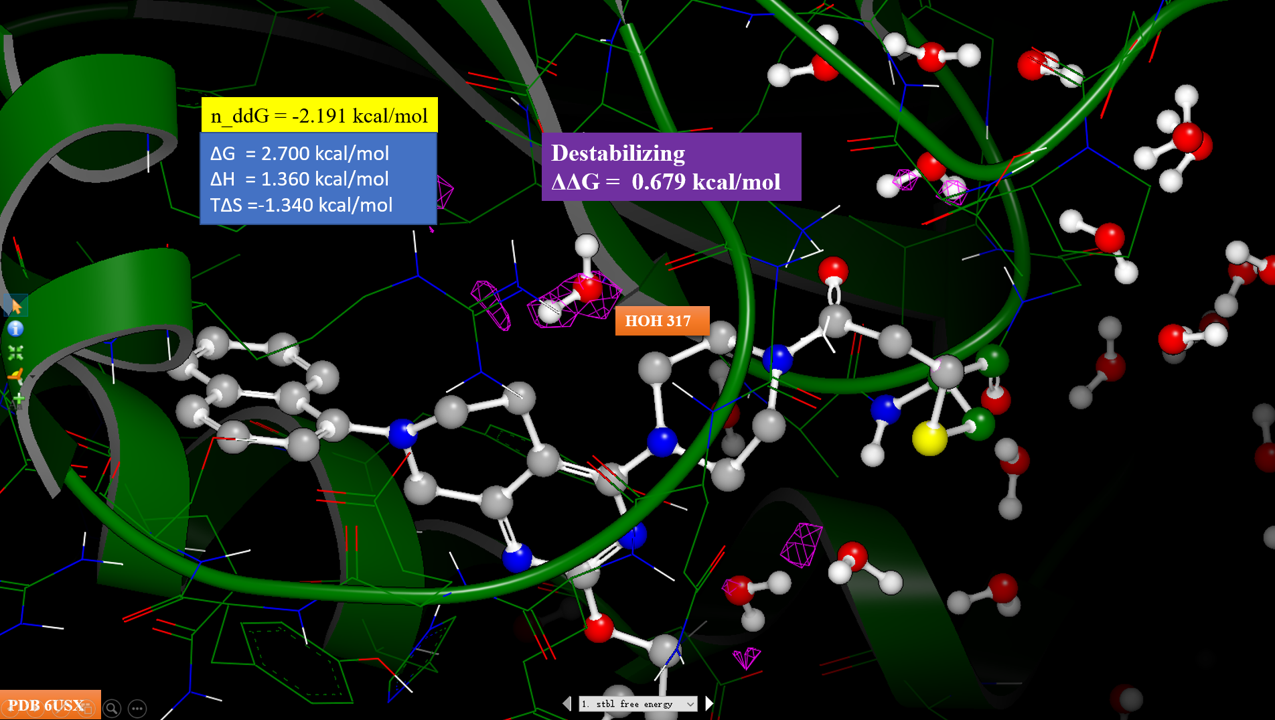
图12. 化合物7与KRAS的共晶结构(PDB 6USZ)的稳定化自由能计算结果。紫色等值图:ΔΔG大于0.5kcal/mol区;黄色等值图:ΔΔG小于-0.5kcal/mol区。
对化合物7与KRAS共晶结构PDB 6USZ进行稳定化自由能计算,并将稳定化自由能表示为等值图,如图9所示,紫色等值图:ΔΔG大于0.5kcal/mol区;黄色等值图:ΔΔG小于-0.5kcal/mol区。我们可以发现,HOH317处于正稳定化自由能区,这意味着HOH317对化合物7与KRAS的结合不利,应该予以替换或修饰配体以改善与水的相互作用。
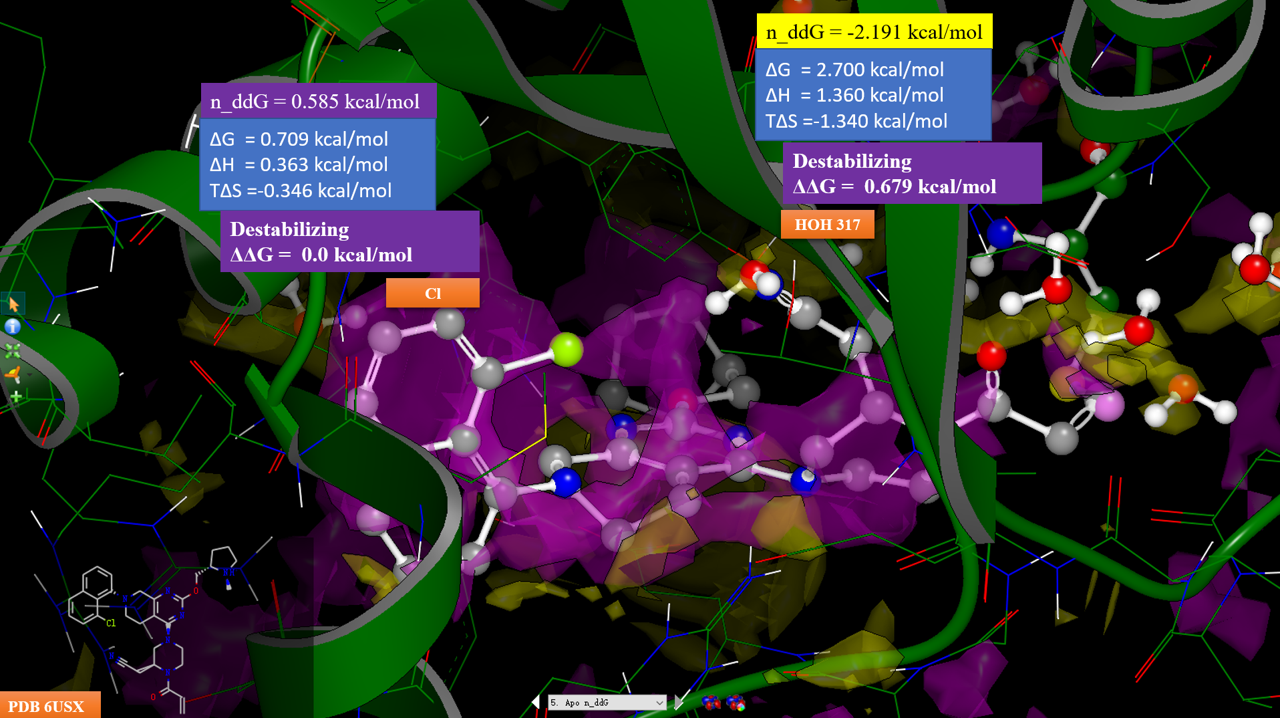
图13. 化合物7-KRAS复合物结构(PDB 6USZ)Apo结构的中性探针自由能差计算结果。紫色等值图:n_ddG大于0.5kcal/mol区;黄色等值图:n_ddG小于-0.5kcal/mol区。配体为化合物9,其构象是将PDB 6UT0按CΑ叠合到PDB 6USX之后修改其中的配体MTRX849而来。
对6USX的Apo结构(不包含配体、水、金属)进行中性探针自由能差分析,等值图分布如图13所示,紫色为配体极性基团不利于活性区域,黄色为配体极性基团有利于活性区域,反之相反。可以发现,HOH317水被一个黄色的区块覆盖,且n_ddG = -2.191 kcal/mol,这说明该位点应该用一个极性基团对HOH317进行替换。为了验证这一点,将MTRX849与KRAS的复合物结构PDB 6UT0叠合到6USX上,并将6UT0配体的氟原子修改氢而得到化合物9的构象。叠合之后,我们可以发现,哌嗪环上的乙腈基氮原子与水叠合在一起。因此我们可以确认,化合物8、9的腈基对HOH317的替换是其实现活性提升440倍的关键。
从图13还可以看到,化合物9的氯原子刚好落在紫色的区块里,氯原子位置对应的n_ddG = 0.585 kcal/mol,这说明氯是靠疏水效应踢走“可能”的水而在活性上获益,化合物9因此比8的活性提高10倍。实际上,根据Fell等人[6]的报道,在8位引入三氟甲基、甲基、乙基等疏水基团都是对活性有利的,而引入腈基与甲氧基则对活性不利,这进一步证明了n_ddG给出的SAR信息的正确性,
GamePlan:将水的能量学信息转化为配体结构修饰假设,指导分子设计
GamePlan应用程序通过对共晶或对接得到的蛋白-配体复合物结构进行分析以确定可能令人感兴趣的溶剂信息位置,并在这些位置进行SZMAP计算,然后处理结果,为配体的修饰提出各种假设并识别水稳定或不稳定的位点。
用VIDA的WaterColor Extension可以为GamePlan的计算结果及其对结构优化的修饰假设提供直观的注释:
- 黄色网状球:极性取代对活性有利位点,球的大小表示极性相互作用的大小。
- 绿色网状球:VDW取代对活性有利位点, 球体的大小表示 VDW 相互作用的大小。
- 绿色半透明球:非极性取代对活性有利位点。
- 紫色:错配位点,配体的极性与溶剂极性不匹配。
- 绿色和红色小球体:分别是起到稳定和去稳定的水合位点。
Murray等人[8]报道了片段筛选发现的HSP90抑制剂苗头化合物10(图14 左),但是它的活性很低,IC50 = 790 μM。经过优化,在苯环3位的甲氧基用疏水性更强的异丙基替换,并在6位引入亲水的羟基,得到的新化合物11(图14 右)的活性提高到14100倍,IC50 = 56 nM。

图14. Murray等人[8]用片段筛选发现的HSP90抑制剂苗头化合物10(IC50 = 790 μM)以及优化的化合物11(IC50 = 56 nM)。
用GamePlan分析化合物10与Hsp90的共晶结构(PDB 2XDL),可以得到三组SAR信息:
- 将配体与结合位点中的溶剂在蛋白背景中进行比较分析,在配体原子周围用彩色注释表明位点类型(图15)。
- 提出对配体进行取代的一系列假设。如图16与17所示,每个取代基在VIDA中以显示为粉红色的虚拟原子,并带有指示位点类型的注释:极性(polar)、非极性(nonpolar)和VDW(van der Walls)。
- 给出起到稳定化或去稳定化作用的水合位点,如图18所示。
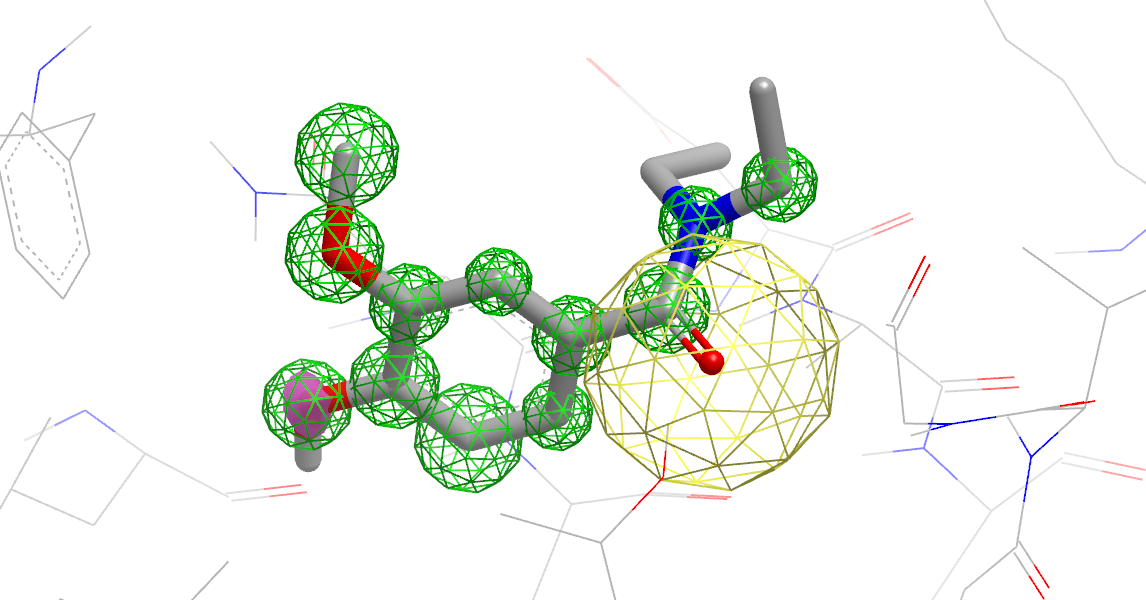
图15. 配体与结合位点中的溶剂在蛋白背景中进行比较分析
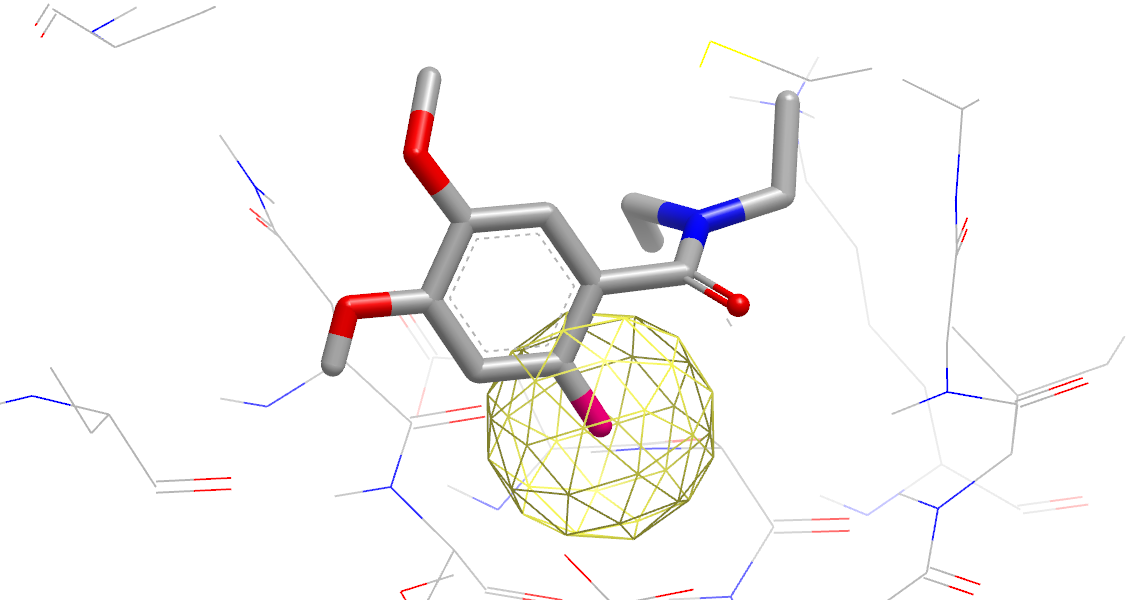
图16. 极性取代对活性有利的假设
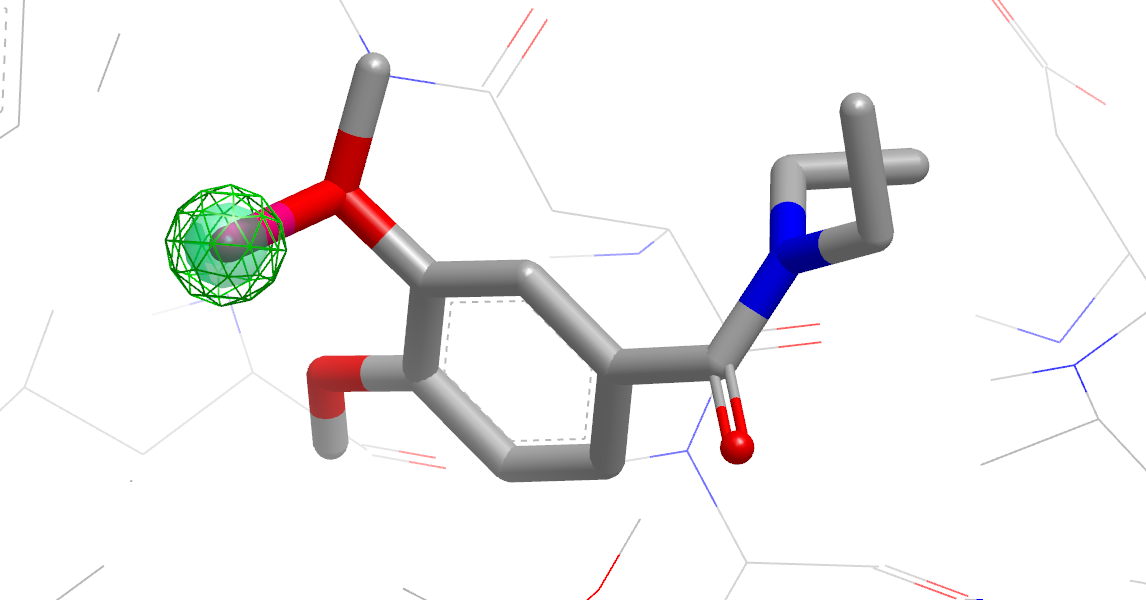
图17. 非极性或VDW取代对活性有利的假设
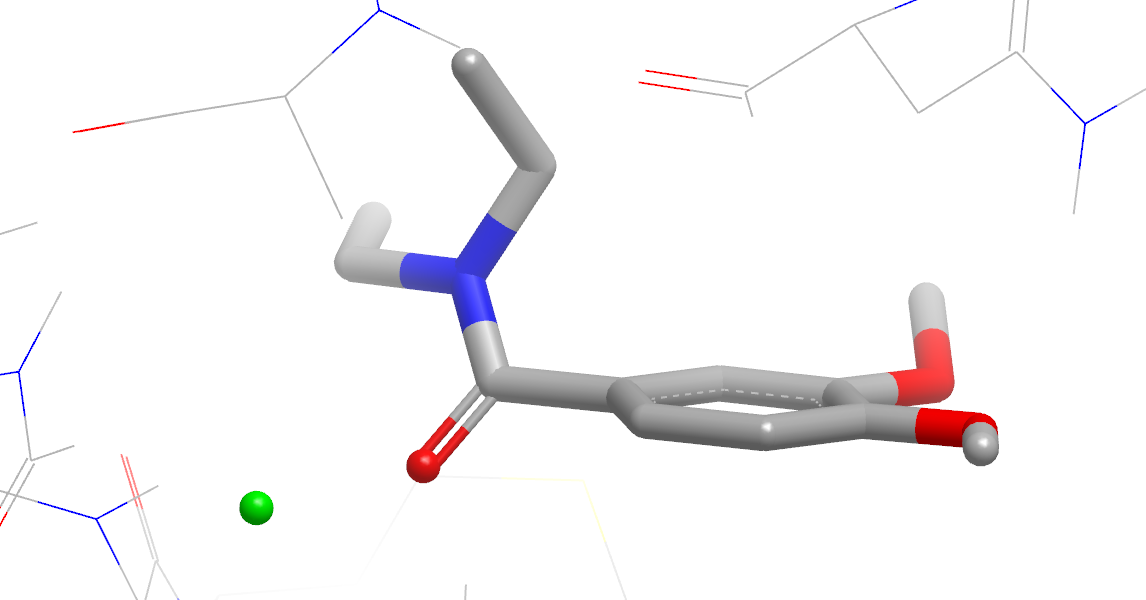
图18. 稳定化与去稳定化水合位点
将上面的GamePlan计算结果信息串联在一起,可以重现从苗头化合物10到11的优化过程,得到如图19所示的结构修饰假设(中间):黄色网格球意味着引入极性取代基团对活性有利;实心浅绿色球与网格状球意味着引入非极性取代或VDW相互作用取代基团对活性有利。显而易见地,化合物11是结构修饰假设的一个完美实现,最终实现从苗头化合物10到11活性14100多倍的提升。

图19. GamePlan分析10与Hsp90共晶复合物(PDB 2XDL)得到的结构修饰假设及优化结果
文献
- SZMAP (Version: 1.6.2.1). https://docs.eyesopen.com/applications/szmap
- Tanger, J. C.; Pitzer, K. S. Calculation of the Thermodynamic Properties of Aqueous Electrolytes to 1000.Degree.C and 5000 Bar from a Semicontinuum Model for Ion Hydration. J. Phys. Chem. 1989, 93 (12), 4941–4951. https://doi.org/10.1021/j100349a053.
- Rashin, A. A.; Bukatin, M. A. Continuum-Based Calculations of Hydration Entropies and the Hydrophobic Effect. J. Phys. Chem. 1991, 95 (8), 2942–2944. https://doi.org/10.1021/j100161a002.
- Grant, J. A.; Pickup, B. T.; Nicholls, A. A Smooth Permittivity Function for Poisson-Boltzmann Solvation Methods. J. Comput. Chem. 2001, 22 (6), 608–640. https://doi.org/10.1002/jcc.1032.
- Kung, P. P.; Sinnema, P. J.; Richardson, P.; Hickey, M. J.; Gajiwala, K. S.; Wang, F.; Huang, B.; McClellan, G.; Wang, J.; Maegley, K.; et al. Design Strategies to Target Crystallographic Waters Applied to the Hsp90 Molecular Chaperone. Bioorganic Med. Chem. Lett. 2011, 21 (12), 3557–3562. https://doi.org/10.1016/j.bmcl.2011.04.130.
- Nittinger, E.; Gibbons, P.; Eigenbrot, C.; Davies, D. R.; Maurer, B.; Yu, C. L.; Kiefer, J. R.; Kuglstatter, A.; Murray, J.; Ortwine, D. F.; et al. Water Molecules in Protein–Ligand Interfaces. Evaluation of Software Tools and SAR Comparison. J. Comput. Aided. Mol. Des. 2019, 33 (3), 307–330. https://doi.org/10.1007/s10822-019-00187-y.
- Fell, J. B.; Fischer, J. P.; Baer, B. R.; Blake, J. F.; Bouhana, K.; Briere, D. M.; Brown, K. D.; Burgess, L. E.; Burns, A. C.; Burkard, M. R.; et al. Identification of the Clinical Development Candidate MRTX849 , a Covalent KRAS G12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63 (13), 6679–6693. https://doi.org/10.1021/acs.jmedchem.9b02052.
- Murray, C. W.; Carr, M. G.; Callaghan, O.; Chessari, G.; Congreve, M.; Cowan, S.; Coyle, J. E.; Downham, R.; Figueroa, E.; Frederickson, M.; et al. Fragment-Based Drug Discovery Applied to Hsp90. Discovery of Two Lead Series with High Ligand Efficiency. J. Med. Chem. 2010, 53 (16), 5942–5955. https://doi.org/10.1021/jm100059d.