QM/MM教程:AMBER-Gaussian联用进行QM/MM模拟
一、声明
本教程转自(2015年1月15日获取):http://sf.anu.edu.au/collaborations/amber_on_fujitsu/amber-12/tutorial/amber-gaussian/index.html
二、教程简介
本教程展示了如何用AMBER12与Gaussian(03或09)进行QM/MM模拟,包括在QM与MM部分切断一个共价键。
三、教程目标
用AMBER-Gaussian界面建立、运行QM/MM模拟作业。
四、预先准备
- Gaussian (g09 or g03)
- Amber 12 (sander, tleap/xleap)
- Amber 12 manual
- 任意一种文本编辑器,比如vi等
- 其它可选的软件: 分子可视化程序
五、操作步骤
本教程包含如下操作步骤
- 用tleap搭建组氨酸3D分子与AMBER计算所需的拓扑与3D坐标文件
- sander的QM/MM计算作业文件的建立
1,用tleap搭建组氨酸3D分子与AMBER计算所需的拓扑与3D坐标文件
首先,我们用tleap或xleap搭建一个小分子–组氨酸用于QM/MM计算。
A. 打开tleap
$ tleap -s -f leaprc.ff12SB
I: Adding /apps/amber/12/dat/leap/prep to search path.
-I: Adding /apps/amber/12/dat/leap/lib to search path.
-I: Adding /apps/amber/12/dat/leap/parm to search path.
-I: Adding /apps/amber/12/dat/leap/cmd to search path.
-s: Ignoring startup file: leaprc
-f: Source leaprc.ff12SB.
Welcome to LEaP!
Sourcing: /apps/amber/12/dat/leap/cmd/leaprc.ff12SB
Log file: ./leap.log
Loading parameters: /apps/amber/12/dat/leap/parm/parm10.dat
Reading title:
PARM99 + frcmod.ff99SB + frcmod.parmbsc0 + OL3 for RNA
Loading parameters: /apps/amber/12/dat/leap/parm/frcmod.ff12SB
Reading force field modification type file (frcmod)
Reading title:
ff12SB protein backbone and sidechain parameters
Loading library: /apps/amber/12/dat/leap/lib/amino12.lib
Loading library: /apps/amber/12/dat/leap/lib/aminoct12.lib
Loading library: /apps/amber/12/dat/leap/lib/aminont12.lib
Loading library: /apps/amber/12/dat/leap/lib/nucleic12.lib
Loading library: /apps/amber/12/dat/leap/lib/ions08.lib
Loading library: /apps/amber/12/dat/leap/lib/solvents.lib
B.建立组氨酸分子
> his=sequence {ACE HIS NME}
> desc his
UNIT name: ACE
Head atom: null
Tail atom: null
Contents:
R<ACE 1>
R<HIE 2>
R<NME 3>
C.将3D文件保存为PDB格式(可以用视图软件查看)
> savepdb his his-ini.pdb
Writing pdb file: his-ini.pdb
D.保存分子的拓扑与3D坐标文件给sander使用
> saveamberparm his his.top his.crd
Checking Unit.
Building topology.
Building atom parameters.
Building bond parameters.
Building angle parameters.
Building proper torsion parameters.
Building improper torsion parameters.
total 8 improper torsions applied
Building H-Bond parameters.
Not Marking per-residue atom chain types.
Marking per-residue atom chain types.
(no restraints)
E.退出tleap
> quit
Quit
下图是我们建立的分子结构:

如你所见,有两个氢原子挨着很近(steric clash),这个结构远不理想。我们打算用Gaussian的QM对侧链部分进行处理而其余部分用分子力场处理(QM/MM Interface处两边分别用不同方法处理)。
2.sander计算QM/MM的参数文件
QM/MM体系的分配与模拟用AMBER12的sander来进行。sander的输入文件分三部分:
- &cntrl部分激活QM计算
- &qmmm部分控制QM/MM模拟
- &gau定义Gaussian计算参数
A. &cntrl部分激活QM计算
首先,&cntrl包含有模拟的通用选项。用namelist变量ifqnt = 1来激活QM计算。
&cntrl
imin=1, maxcyc=10, (Perform minimization for 10 steps)
ntb=0, (Non-periodic simulation)
cut=20., (20 angstrom classical non-bond cut off)
ifqnt=1 (Switch on QM/MM coupled potential)
&end
B.&qmmm部分控制QM/MM模拟
&qmmm含有控制QM/MM模拟的选项。QM/MM选项必须定义该部分的iqmatoms 或 qmmask 变量:它们定义了QM处理的区域。
&qmmm
qmmask=’@11-21′ (这些原子用QM处理)
qmcharge=0, (QM区电荷为0)
qm_theory=’EXTERN’, (必须设置以激活,must be set to enable the external interface)
qmcut=20.0 (Use 20 angstrom cut off for QM region)
&end
变量qm_theory = ‘EXTERN’是必须的,用来激活QM/MM计算调用外部程序。
QM处理的原子数取自之前保存的PDB文件。
C. &gau部分控制Gaussian计算参数
这部分可以是 &adf, &gms, &nw, &gau, &orc or &tc分别调用ADF, GAMESS, NWChem, Gaussian, ORCA 或TeraChem进行计算,并为外部QM程序设定参数。本例我们用Gaussian来计算,所以用&gau。
&gau
method = BLYP, (Method to be used in the calculation)
basis = 6-31G, (Basis set type to be used in the calculation)
charge = 0 (Net total charge of the QM region)
&end
全部的 &gau namelist 变量:
| basis | Basis set type to be used in the calculation. Any basis set that is natively supported by Gaussian can be used. Examples are the single zeta, split valence or triple zeta Pople type basis sets STO-3G, 3-21G, 6-31G and 6-311. The split-valence or triple zeta basis sets can be augmented with diffuse functions on heavy atoms or additionally hydrogen by adding one or two plus signs, respectively, as in 6-31++G. Polarization functions on heavy atoms or additionally hydrogens are used by adding one or two stars, respectively, as in6-31G. (Default: basis = 6-31G*) |
| method | Method to be used in the calculation. Can either be one of the WFT models for which Gaussian supports gradients, for example RHF or MP2, or some supported DFT functional. Popular choices are BLYP, PBE and B3LYP. (Default: method = BLYP) |
| scf_conv | Threshold upon which to stop the SCF procedure. The tested error is the commutator of the Fock matrix and the density matrix. Convergence is considered to be achieved if the maximum element of the commutator (which is zero for an optimized wave function) is smaller than scf_conv}. Set in the form of 10-N. (Default: scf_conv = 8) |
| charge | Net total charge of the QM region. (Default: charge = 0) |
| spinmult | Net total spin multiplicity of the QM region. Open shell systems (spin multiplicity larger than 1) are treated with unrestricted HF, MP2, or KS approaches. (Default: spinmult = 1) |
| num_threads | Number of threads (and thus CPU cores) for Gaussian to use. Unless num_threads is explicitly specified, Gaussian will only use one thread (run on one core). (Default: num_threads = 1) |
| use_template | Determine whether or not to use a user-provided template file for running external programs. (Default: use_template = 0) |
| ntpr | Controls frequency of printing for dipole moment and atomic charges to files gau_job.ext. (Defaults to &cntrl namelist variable ntpr) |
| dipole | Toggles reading/printing for dipole moment. (Default: dipole = 0) |
本例子中,QM/MM计算所用的完整的sander输入文件如下:
Example QM/MM input for Sander-12 - Gaussian interface &cntrl imin=1, maxcyc=10, ntb=0, cut=20., ifqnt=1 &end &qmmm qmmask='@11-21' qmcharge=0, qm_theory='EXTERN', qmcut=20.0 &end &gau method = BLYP, basis = 6-31G, charge = 0 &end
将之保存为amber-g09-min.in, 现在已经准备好开始跑sander作业了。
提交sander作业
可以开始跑sander了,键入如下命令:
$ sander -O -i amber-g09-min.in -o amber-g09-min.out \
-c his.crd -p his.top -r amber-g09-min.rst
如果是大的计算,提交给PBS进行计算(比如, run.sh)。
检查结果
计算完后,检查目录。可以看到几个新文件:
$ ls -l total 1000 -rw-r----- 1 vvv900 z01 268 Jul 12 15:38 amber-g09-min.in -rw-r----- 1 vvv900 z01 11431 Jul 12 15:46 amber-g09-min.out -rw-r----- 1 vvv900 z01 1070 Jul 12 15:46 amber-g09-min.rst -rw-r----- 1 vvv900 z01 1 Jul 12 15:46 gau_job.chk -rw-r----- 1 vvv900 z01 1951 Jul 12 11:02 his-ini.pdb -rw-r----- 1 vvv900 z01 1070 Jul 12 11:03 his.crd -rw-r----- 1 vvv900 z01 25840 Jul 12 11:03 his.top -rw-r----- 1 vvv900 z01 17501 Jul 12 11:13 leap.log -rw-r----- 1 vvv900 z01 412 Jul 12 15:46 mdinfo -rw-r----- 1 vvv900 z01 3663 Jul 12 15:46 old.gau_job.inp -rw-r----- 1 vvv900 z01 47302 Jul 12 15:46 old.gau_job.log -rw-r----- 1 vvv900 z01 277 Jul 12 15:28 run.sh
amber-g09-min.out 是sander的输出文件。
将QM部分从蛋白截出会使得MM部分的总电荷不是一个整数,虽然如此,sander会自动的进行调整:
Forcing neutrality...
QMMM: ADJUSTING CHARGES
QMMM: ----------------------------------------------------------------------
QMMM: adjust_q = 2
QMMM: Uniformly adjusting the charge of MM atoms to conserve total charge.
QMMM: qm_charge = 0
QMMM: QM atom RESP charge sum (inc MM link) = -0.022
QMMM: Adjusting each MM atom resp charge by = -0.001
QMMM: Sum of MM + QM region is now = 0.000
QMMM: ----------------------------------------------------------------------
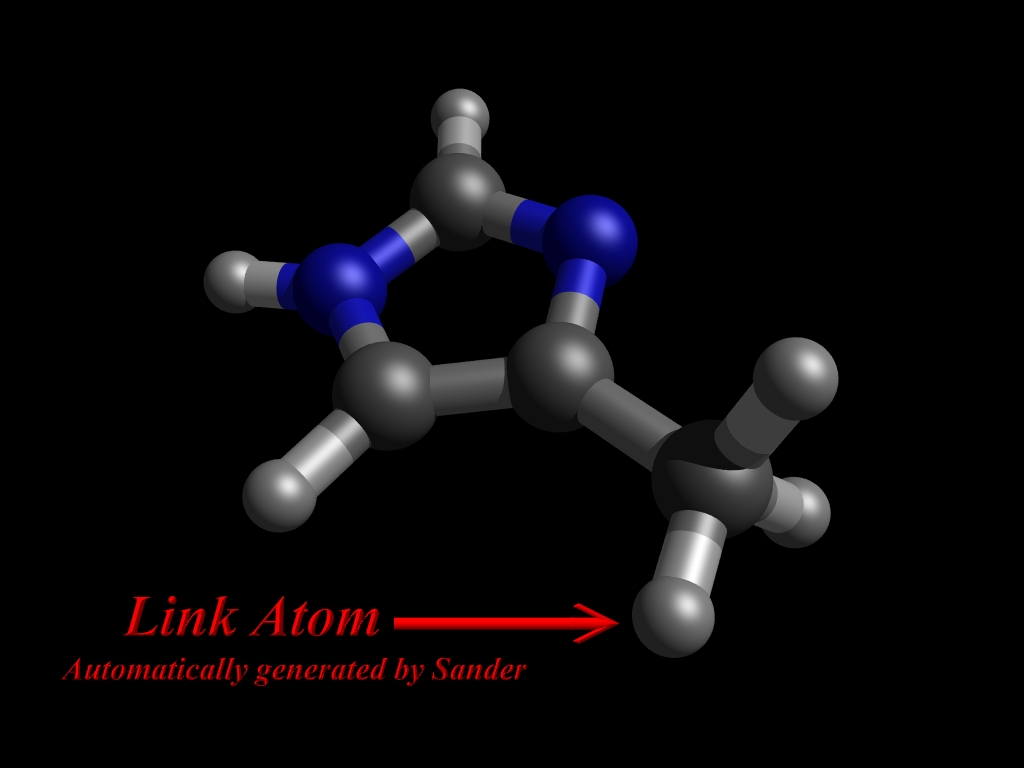
Then sander informs us about about a covalent bond between the QM and MM parts which should be cut:
QMMM: Link Atom Information
QMMM: ------------------------------------------------------------------------
QMMM: nlink = 1 Link Coords Resp Charges
QMMM: MM(typ) - QM(typ) X Y Z MM QM
QMMM: 9 CX 11 CT 5.084 4.502 -0.351 -0.058 -0.007
QMMM: ------------------------------------------------------------------------
然后sander会输出QM原子的坐标文件,包括自动为链接原子生成质子。
QMMM: QM Region Cartesian Coordinates (*=link atom)
QMMM: QM_NO. MM_NO. ATOM X Y Z
QMMM: 1 11 C 5.6613 4.2208 -1.2321
QMMM: 2 12 H 5.8085 3.1409 -1.2414
QMMM: 3 13 H 5.1233 4.5214 -2.1312
QMMM: 4 14 C 7.0335 4.8447 -1.3211
QMMM: 5 15 N 7.8926 4.5867 -2.3830
QMMM: 6 16 C 9.0051 5.2681 -2.1825
QMMM: 7 17 H 9.8193 5.2069 -2.9046
QMMM: 8 18 N 8.8989 5.9379 -1.0617
QMMM: 9 19 H 9.6126 6.5402 -0.6769
QMMM: 10 20 C 7.6863 5.7028 -0.4925
QMMM: 11 21 H 7.4253 6.1816 0.4513
QMMM: 12 *H 5.0838 4.5018 -0.3515
我们还可以检查一下能量分解部分:
NSTEP ENERGY RMS GMAX NAME NUMBER
1 -1.6571E+05 1.2411E+03 7.1568E+03 H 25
BOND = 0.0206 ANGLE = 0.4312 DIHED = 16.9792
VDWAALS = 792.1448 EEL = -82.0092 HBOND = 0.0000
1-4 VDW = 5.5589 1-4 EEL = 44.7113 RESTRAINT = 0.0000
EXTERNESCF = -166492.6739
我们看到,QM/MM结果有一行为EXTERNESCF。这是在MQ/MM计算QM部分的能量。
其它的文件包括:
- gau_job.chk 是Gaussian checkpoint file
- mdinfo 是mdout格式的能量信息文件
- old.gau_job.inp 是Gaussian输入文件,自动的有sander生成
- old.gau_job.log 是Gaussian输出文件
检查一下优化过的结构,发现立体碰撞不见了:

检查Gaussian输出文件,我们会发现QM区被自动加上H原子作为链接原子(link atom)。
六,相关文件下载
本附件包含了组氨酸的拓扑文件与3D结构文件,sander的输入文件与输出文件等,下载:http://www.molcalx.com.cn/wp-content/uploads/2015/01/qm_mm_tutorial.zip
七,联系我们
工作时间:工作日9:30-17:30
公司电话:020-38261356
公司传真:4008892163转519265
公司邮箱:info@molcalx.com
公司地址:广州市天河区龙口东路34号龙口科技大厦20楼2002室
邮 编:510630
微信号:molcalx
新浪/腾讯微博:molcalx